(RIP1)/RIP3-Regulated Necroptosis, Interplay with Apoptosis and Autophagy, and Its Therapeutic Effect on Leukemia
*Corresponding Author(s):
Malak Yahia M QattanDepartment Of Health Science, College Of Applied Medical Science And Community Services, King Saud University, Riyadh, Saudi Arabia
Tel:+966 556333337,
Email:mqattan@ksu.edu.sa
Abstract
Apoptosis resistance is a major obstacle leading to chemotherapy failure during cancer treatment. Necroptosis is an alternative mode of programmed cell death with necrotic morphology, mediated by signal transduction from Receptor-Interacting serine/threonine kinase (RIP1) to RIP3 and its modality is the caspase-independent pathway. Necroptosis cell death overcomes apoptosis resistance and amplifies antitumor immunity in cancer therapy. The role of necroptosis in leukemia is unknown. This mini review aims to provide a better understanding of necroptotic pathway mechanisms, their interaction with other forms of cell death, and the implications of necroptosis and RIP-1/RIP-3 on leukemia treatment. First, a brief introduction of the types of cell death is given. The interplay between necroptosis and other cell death mechanisms in leukemia is then explained and a summary of the effects of necroptosis on leukemia therapeutics is provided.
Keywords
Apotosis; Autophagy; Necroptosis, Leukemia, Receptor-interacting serine/threonine kinase (RIP1) to RIP3
ABBREVIATIONS
ALL: Acute Lymphoblastic Leukemia
AML: Acute Myeloid Leukemia
FADD: Fas-Associated protein with Death Domain
HDAC: Histone Deacetylase
Nec- 1: Necrostatin-1
NF-κB: Nuclear Factor kappaB
RIP1/RIP3: Receptor-interacting serine/ threonine-protein
Smac: Second mitochondria-derived activator of caspases
TNF: Tumor Necrosis Factor
TNFR: TNF Receptor
Bcl-2: B cell lymphoma-2
BH-3: Bcl-2-Homology
Cyt c: Cytochrome c
DD: Death Domain
IAP: Inhibitor of Apoptosis Protein
JNK: Jun N-terminal Kinase;
LC3: Microtubule-associated protein 1A/1B-light chain 3
TRADD: TNF Receptor-Associated Death Domain
VDAC: Voltage-Dependent Anion Channel
MAPK: Mitogen-Activated Protein Kinases
PI3K: Phosphatidyl Inositol 3-Kinase;
PE: Phosphatidyl Ethanolamine
SMAC: Second Mitochondria derived Activator of Caspase
APAF- 1: Apoptosis Protease Activating Factor
MAC: Mitochondrial outer membrane
TRAIL: TNF-Related Apoptosis-Inducing Ligand
LC3-II: Phosphatidylethanolamine conjugate
RHIM: RIP1 Homotypic Interaction Motif
cIAPs: cellular Inhibitor of Apoptosis Protein
MLKL: Mediator Mixed-Lineage Kinase Domain like.
INTRODUCTION
Cell death is the stopping of a biological cell from functioning. This may be the result of a normal process in which old cells die and are replaced by new ones, or may be caused by other factors such as diseases, injury to a specific organ, or death of the organism itself and considerable importance in the cell’s life cycle [1].
Dead cells can be identified based on morphological or molecular features that include loss of cell membrane integrity, fragmentation of cell organelles such as the nucleus, and phagocytosis of cell fragments [2]. Cell death can be categorized as autophagic, necrotic, and apoptotic, depending on the cell morphology (Figure 1) [2,3].
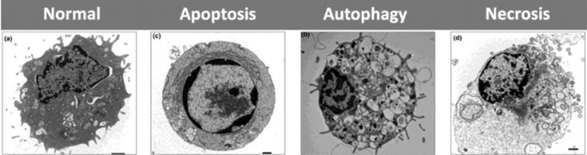
Figure 1: Shows the morphological features and differences between apoptosis, autophagy and necrosis versus normal cell. a) Normal cell; b) autophagic showing the massive formation of (double-membraned) autophagic vacuoles; c) apoptotic cell showing the typical round shape and chromatin condensation; d) necrotic cell showing increased cell size, rupture of cell membrane and vacuoles formation.
PROGRAMMED CELL DEATH (APOPTOSIS)
In a healthy adult human being, around 10 million cells die via apoptosis every day. Apoptosis is mostly described as programmed cell death since it is a phenomenon during which a cell sets off its own death i.e. suicide [4]. The degree of apoptotic death is important in maintaining homeostasis, as well as, in controlling the shape and size of tissues during various developmental processes. This programmed cell death is also essentially required to reduce the number of immune effector cells once the pathogen has been eliminated [5].
During apoptosis, a cell becomes round, and its volume is reduced. This process is called pyknosis. In addition, chromatin material becomes condensed and the nucleus becomes fragmented. This phenomenon is called karyorrhexis. Apoptosis is also accompanied by plasma membrane blebbing. However, there occur insignificant ultra-structural modifications of cellular organelles. Blebs serve as the progenitors of apoptotic bodies having small, almost round cytoplasmic fragments surrounded by cell membrane. Moreover, it is possible that apoptotic bodies contain functional organelles enclosed in unbroken cell membrane [6,7]. Apoptotic bodies express phosphatidylserine on their surface. It is a phospholipid attached to the cell membrane and it functions as an “eat me” signal in order to attract phagocytic cells such as macrophages, which phagocytose these bodies [1,6,7].
There are two main pathways to apoptosis, namely, the death receptor pathway (extrinsic pathway) and the intracellular stress signals pathway (intrinsic pathway) [8].
The extrinsic pathway is activated by signals such as toxins, hormone, growth factors, nitric oxide or cytokines. The signals come from outside the cell and transduce through the plasma membrane for apoptosis to occur. The intrinsic pathway initiates in response to glucocorticoid and signals such as heat, radiation, hypoxia, viral infection, or raised calcium level inside the cell [9].
Apoptosis extrinsic pathway (Death receptor pathway)
Tumour Necrosis Factor (TNF) is the cytokine generated from activated macrophages and initiates the apoptosis through the extrinsic pathway [9]. TNF combines with Tumour Necrosis Factor-Receptor (TNF-R) and subsequently activates caspase, a fundamental apoptotic protein. The intermediate membrane protein called TNF Receptor-Associated Death Domain (TRADD) and Fas-Associated Death Domain protein (FADD) signal to caspase for apoptosis [8].
Apoptosis intrinsic pathway
The transduction of intrinsic pathway inside the cell can increase permeability of mitochondrial outer membrane, thus, allowing mitochondrial proteins to escape and disperse into the cell cytosol [10].
Mitochondria are the most crucial organelles in the regulation of apoptosis. Cytochrome c (Cyt c) is the first protein released from mitochondria for apoptosis, through configurative channels on mitochondrial outer membrane (MAC). Cyt c binds to apoptosis protease activating factor (APAF-1) to form Cyt c/APAF-1 complex termed as apoptosome [11]. Cyt c/APAF-1 complex consequently binds to pro-caspase-9; then apoptosome cleaves pro-caspase into active form with the purpose of activating caspase 3 [10].
Voltage Dependent Anion Channel (VDAC) is a mitochondrial outer membrane protein that can control apoptosis by regulating mitochondrial outer membrane pores permeability [9]. The second mitochondria derived activator of caspase (SMAC) is a regulatory protein released in the cell, as a result of an increase in mitochondrial permeability. It plays an important role in apoptosis, as it deactivates inhibitor of apoptosis protein (IAP) which represses caspase activity [7-9]. Smac/Diablo is the second apoptotic factor after Cyt c but with a different function. Cyt c is the direct trigger of APAF-1 and caspase-9 whereas Smac/Diablo antagonize Baculovirus Inhibitor Repeat (BIR - a domain of IAP) and facilitates activation of caspase indirectly [9].
AUTOPHAGY
About fifty years ago, researchers discovered the phenomenon of autophagy. However, substantial molecular information about this process has been obtained during the past 5-7 years. This information has given insight into the form and functions of this phenomenon [12,13]. It has now been established that autophagy is the major cellular catabolic mechanism that safeguard the cell from different stresses such as deficiency of growth factors, nutrients or hypoxia instigated stress. It plays a crucial role in recycling the cellular organelles, proteins and other components. Therefore, it ensures cleanup of the cell, as well as, the provision of new constituents to build up various parts of the cell [14]. Autophagy is an ubiquitous phenomenon that take place in all types of cells. However, deficiency of nutrients or other stresses can result in upregulation of this process [14]. Researchers have also determined an association between autophagy and etiology of a number of human diseases such as cancer, metabolic disorders and neurodegenerative disorders [15].
Autophagy is of substantial importance for humans, especially in protection from a number of disorders. Its role in causation and prevention of different disorders is now well established. Numerous myodegenerative and neurodegenerative disorders are described by an elevated, yet inadequate autophagic activity. This shows how important autophagy is for the quality control of proteins. Likewise, if protein damage, mitochondrial damage or DNA damage caused by cellular stress is not followed by appropriate autophagic repair pathways, the affected cells become susceptible to becoming cancerous cells [16]. As mentioned above, induction of autophagy occurs in response to different cellular stress conditions like nutrient deficiency. Autophagosomes which are intracellular organelles and proteins impounded by double membrane vesicles are delivered to lysosomes during this process, so that they can be degraded. A growing number of evidence indicates that autophagy acts as a membrane trafficking system which supplies the parts of cytoplasm to the lysosomes for degrading protein in bulk, thereby, ensuring not only the survival of the cells but also quality control of the protein content of the cell through the elimination of abnormal proteins. Even though autophagy is viewed as a pathway for ensuring survival of the cell, unnecessary autophagy can induce necroptosis i.e., the so called type II programmed cell death.
MECHANISM OF AUTOPHAGY
During the course of autophagy, an autophagosome is formed when phagophore closes after engulfing cytoplasmic constituents and cellular organelles. Phagophore is a pre-autophagosomal structure having acup-like shape and is also known as isolation membrane [17]. The next step is normally a fusion of autophagosome with lysosomal vesicles, resulting in proteolytic breakdown of the engulfed substances through the action of lytic enzymes contained in the lysosomes [18].
A pair of ubiquitin-like modifications is needed for the preliminary development of the sequestering membrane. These include the formation of Atg12-Atg5 conjugation system and the Atg8-Phosphatidylethanolamine (PE) complex [19]. In Atg12-Atg5 conjugation system, a covalent bond binds the smaller Atg12 protein with the Atg5 [20]. Atg7 acts as E1 enzyme and activates the Atg12 first, followed by the development of thioester bond between the two proteins. Then, Atg12 gets linked to Atg10 which is E2-like enzyme, resulting in the formation of a new thioester bond. Finally, through its glycine at carboxyl end, the Atg12 protein gets attached to amino group in the lysine residue of Atg5 [20,21]; however, this bonding is irreversible. Through its coiled coil domain at carboxyl end, the Atg16 gets attached to the Atg12-Atg5 complex and this bigger complex forms temporary association with the isolation membrane or phagophore [19-21]. The second conjugation system needs this complex for its formation. However, the Atg12-Atg5 conjugate splits after the formation of autogphagosome.
The Atg8/LC3-PE complex is the 2nd conjugation system and is the best-defined marker for autophagosomes. Cysteine proteinase Atg4 processes Atg8 by cleaving the arginine residues at the carboxyl end, thereby exposing a glycine residue. Atg7 activates this glycine followed by transference of Atg8, an E2-lke enzyme, to Atg3 [20]. Eventually, the conjugation of Atg8 with phospholipids PE occurs, thus, permitting the binding of protein with the autophagosomal membrane. The Atg8-PE complex is dissociated by the same Atg4 enzyme, leading to the recycling of Atg8 back to the cytoplasm. The cytosolic LC3-I form is produced when an Atg4 cysteine proteinase i.e. autophagin cleaves the newly formed LC3. The LC3-I form, having molecular weight of 18kDa, undergoes few ubiquitination-like reactions, leading to the generation of the membrane bound protein LC3-II. This LC3-II protein (16kDa) binds to both the outer and the inner sides of the developing autophagosome [22].
AUTOPHAGY SIGNALLING PATHWAY
Autophagy cascade kinases
Phosphatidyl Inositol 3-Kinase (PI3K) is one of the first kinases to be verified as involved in autophagy. There are three groups of PI3K in mammal’s cells [23]. Among these groups, class III PI3K plays an important role in the authophagy mechanism. Class III PI3K activates autophagy. On the other hand, autophagy is negatively regulated by class I PI3K. It has also been established that class III PI3K demonstrates interaction with Beclin 1 and p150. Beclin 1 was actually isolated as Bcl-2 interacting protein. Moreover, it serves as the first autophagy related tumour suppressor gene [24].
Serine/threonine kinase Atg1 is the 2nd major kinase involved in the pathway of autophagy. It develops a complex through interaction with different regulatory proteins and under different nutrient conditions, leading to the formation of complexes with different compositions. Partial dephosphorylation of Atg13 occurs in starvation conditions, followed by development of autophagosome, through an interaction between dephosphorylated Atg13 and Atg1. However, in nutrient-rich environment, phosphorylation of Atg13 occurs and its interaction with Atg1 is inhibited, thereby, leading to inactivation of autophagy. Moreover, under nutrient-deficient conditions, association between Atg17 i.e. another Atg protein, and the Atg1-Atg13 complex is augmented [25].
Autophagy proteins; Beclin 1
Beclin 1 is the first mammalian gene to be identified as having a role in mediating autophagy. Beclin 1 is a novel Bcl-2-homology (BH)-3 domain only protein. Anti-apoptotic Bcl-2 family members interact with the BH3 domain of Beclin 1 [26]. Beclin 1 is localized within the plasma-membrane, the cytoplasm and the nucleus in human and murine [27]. Transcriptionally, nuclear factor (NF)-κB is involved in the regulation of Beclin 1 expression. NF-κB directly binds the Beclin 1 promoter and upregulates its mRNA and protein levels, leading to positive modulation of autophagy in T cells [28].
Autophagy proteins; Microtubule-associated protein 1A/1B-light chain 3 (LC3)
All mammalian tissues and cultured cells possess microtubule-associated protein 1A/1B-light chain 3 (LC3), which is a soluble protein with a molecular weight of about 17 kDa. Various components of cytoplasm such as cellular organelles and proteins found in cytosol are engulfed by autophagosomes during the course of autophagy. At the same time, LC3-I i.e. a cytosolic form of LC3, conjugates with phosphatidylethanolamine, resulting in the formation of LC3-phosphatidylethanolamine conjugate (LC3-II). This conjugate is then recruited to autophagosomal membranes. Autolysosomes are formed by the fusion of autophagosomes and lysosomes. Hydrolase enzymes contained in the lysosomes degrade the intra-autophagosomal components. The degradation of LC3-II in the lumen of autolysosomes occurs simultaneously [29].
PROGRAMMED CELL DEATH II (NECROPTOSIS)
Historically, cell death was believed to be either programmed, called “apoptosis”, or passive, called “necrosis”. Apoptosis was considered instrumental to development and other physiological functions, while necrosis was seen as a response to inflammation conditions. Now, necrosis is known to have programmed aspects, with “necroptosis” describing regulated necrotic cell death that bears a mechanistic features to apoptosis and morphological features to necrosis.
Apoptosis and necrosis are characterized by different mechanisms leading to cell death. Apoptosis is associated with nuclear disintegration, followed by removal of the cell by macrophages. In necrosis, the nucleus stays intact and instead the membrane becomes swelling, resulting in leakage of cellular components. Apoptosis mainly occurs during embryogenesis, while necrosis is more common during disease states. Necroptosis falls somewhere in the middle, with several molecules and processes that can trigger, modulate, and effect necroptosis in cells [30].
Kinnally et al., [31] and Smith and Yellon [32] have both proposed that there are two apoptotic pathways which are partially interlinked. These include the classical apoptosis which is dependent on caspase, and caspase-independent programmed cell death. Both of these mechanisms can be associated with each other, since caspases may cause the activation of non-caspase proteases and vice versa.
Apoptosis is promoted by death receptors through an intracellular motif termed Death Domain (DD), causing the activation of caspases-8 and thereby, inducing apoptosis. Once the caspase-8 is inhibited, activity of the Jun Amino-Terminal Kinase (JNK) and DD-containing kinase RIP-1 results in the induction of a type of cell death which leads to the production of autophagosome, and which is dependent on Atg6/Beclin 1 and Atg7 [26].
It has been recently determined that T lymphocytes, stimulated by mitogens, induce nonapoptotic active capases-8. Moreover, T cells devoid of this caspases-8 activity can enter the cell cycle. Nevertheless, they are unable to accumulate and undergo caspase-independent cell death. It was determined that this cell death results from chemical inhibition of RIP-1 and the necroptotic inhibitor Nec-1 caused restoration of T cells increasing in number. A parallel study conducted by Chen and colleagues reported that knockdown of RIP-1, mediated by Nec-1, restores the ability of T cells lacking in caspase-8, to demonstrate clonal expansion. This implies that kinase activity of RIP-1 is important for this mechanism of cell death [33].
RIP1/RIP3 complex
The human RIP gene encodes seven splicing isoforms: RIP1, RIP2, RIP3, RIP4, RIP5, RIP6 and RIP7 [34]. The Receptor Interacting Protein 1 (RIP1) is essentially required for the induction of necrosis and it is identified to interact with death domain (DD) of receptor Fas (CD95) and trigger programmed death response in cells [35]. Moreover, RIP3 is a protein attenuating RIP1 and tumor necrosis factor receptor 1(TNFR1) induces NK-?B activation [36] RIP1 and RIP3 are regulated by ubiquitination and caspases. NF-κB and Mitogen-Activated Protein Kinases (MAPK) are activated by ubiquitination of RIP1 to enhance cell survival [37]. On the other hand, caspase-8 triggers apoptosis pathway upon deubiquitnation of RIP1 [37]. RIP1/RIP3 complex is formed when RIP1combines with RIP3 via C-terminal death domain, the intermediary RIP-1 Homotypic Interaction Motif (RHIM) as a result of inhibition of caspase-8 [38]. Three different domains can be identified in RIP1. These are the C-terminal death domain, the intermediary RIP1 homotypic interaction motif (RHIM)-domain and the N-terminal kinase domain [39,40]. RIP1/RIP3 complex triggers necroptosis via downstream signal transduction [38].
Upon stimulation of TRAIL or TNF, necrosome is formed, and it in turn, activates RIP3. Upon activation, RIP3 interacts with enzymes regulating glycolytic flux, glutaminolysis, leading to the formation of reactive oxygen species in the mitochondria [39,40]. It has now been established that the activity of RIP1 is linked specifically with necrosis, instead of apoptosis. Moreover, necrostatin-1 (Nec-1), a small molecule which inhibits necroptosis, impedes the RIP1 kinase activity [41]. So far, researchers are unable to identify any substrate of RIP1 that could have a role in necroptosis [1].
A prototypic signaling pathway to necroptosis is engaged by the binding of TNFα to its cognate cell surface receptor TNF receptor 1 (TNFR1), which triggers formation of the so called complex I at the TNFR1 [42]. This leads to K63- linked polyubiquitination of RIP1 by cellular inhibitor Nuclear Factor kappaB (NF-κB). Upon internalization of TNFR1, secondary cell death complexes assemble in the cytosol. When caspase-8 activation is blocked, RIP1 is no longer cleaved and can interact with RIP3 to build up the necrosome resulting in activation of RIP1 and RIP3 in an autocrine/paracrine manner via reciprocal phosphorylation [43]. RIP3 subsequently phosphorylates and activates MLKL, a pseudokinase that lacks intrinsic kinase activity [44]. Upon its activation, MLKL forms oligomers and translocates from the cytosol to the plasma and intracellular membranes, where it disrupts membrane integrity (Figure 2) [45,46].
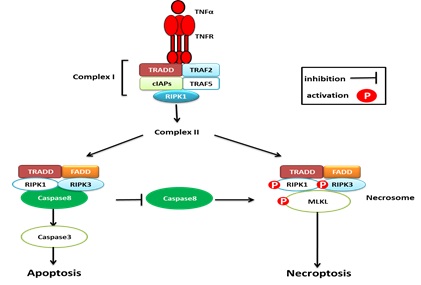
Figure2: TNFα-TNFR1-related signaling pathway of apoptosis and necroptosisin leukemia cells. Upon binding to TNF receptor 1, TNF receptor 1 recruit TRADD, TRAF2 and TRAF5, RIP-1, cIAPs to form complex I. Deubiquitnation of RIPK1 leads to formation of complex II. Caspase-8 activation in complex II leads to apoptosis and suppresses the induction of necroptosis. On the other hand, inactivation of Caspase-8 in complex II leads to the phosphorylation and activation of RIPK1, RIPK3 and MLKL during the assembly of the necrosome to activate necroptosis and suppresses apoptosis.
Necroptosis may offer an alternative option to trigger programmed cell death in cancer cells, as many cancers have evolved mechanisms to evade cell death [47]. In apoptosis-resistant cancers, therapeutic induction of necroptosis may be of particular interest, as activated caspase-8 has been shown to cleave and thus inactivate RIP1 [48]. This implies that apoptosis-resistant cancer cells are particularly susceptible to necroptosis.
LINKING AUTOPHAGY AND APOPTOSIS
There are two possible ways through which one can link the phenomena of autophagy and apoptosis. Firstly, the autophagy could demonstrate a regulatory role for apoptosis by increasing or decreasing its chances to occur. Secondly, apoptosis could demonstrate a regulatory role for autophagy by enhancing or reducing it [13]. Beclin 1 is an autophagic substance which is over expressed under the influence of nutrient depletion, hypoxia, angiogenesis inhibitors, immunotherapy, irradiation, or chemotherapy [49]. Cancerous cells are protected from apoptosis through bid knockdown which also stimulates autophagy and this is manifested by enhanced expression of Beclin 1. However, the current literature is still deficient in elaborating the exact mechanism involved in inhibition of apoptosis by Beclin 1.
During apoptosis, Beclin 1 can be cleaved by caspases, resulting in the confiscation of its pro-autophagic action. Caspases are cysteine aspartyl proteases which play a crucial role in apoptosis and are stimulated by a death receptor ligand i.e., TRAIL [24,50]. Beclin 1 cleavage, mediated by capase-3, -7 and -8, results in the formation of C- and N- terminal fragments, leading to loss of the ability to induce autophagy. The C-terminal fragments move to the mitochondria and this make the cells sensitive to apoptotic signals [51]. Autophagy is decreased by apoptosis triggered by the proapoptotic protein Bax via increasing Beclin 1 cleavage at D149, mediated by caspase [52]. Nevertheless, it has been reported by some latest research studies that autophagy can result in degradation of a death receptor effector i.e. caspase-8, which implies that living systems have a feedback mechanism for cross-regulation of apoptosis and autophagy. Even though autophagy is inactivated by the apoptosis-related cleavage of Beclin 1 and Atg5, caspase-3-mediated Atg4D cleavage produces a fragment, capable of demonstrating enhanced autophagic activity [49].
FUNDAMENTAL NECROPTOSIS MECHANISM IN LEUKAEMIA TREATMENT
Cell death is an essential cellular process thereby justifying the existence of more than one form of cell death processes. Principally, apoptotic pathways that cause cell death are of crucial significance throughout the organism growth and for regulation of the immune system while controlling defense response to a disease stimulus. Cell death is inter-connected with both cell- endurance and cell-expansion, through the control of molecular pathways [53]. It’s evident that inter-connection and inter-dependence among death processes can give diverse outcomes.
Among various types of cell death; apoptosis and autophagy are the most interesting area for researchers. The induction of necroptosis has emerged in recent years as an alternative therapeutic approach to trigger programmed cell death in cancer cells, in particular in apoptosis-resistant cases [54].
It's crucial to investigate the role of necroptosis and RIP1/RIP3 in the combination with apoptosis and autophagy because its role in leukemia and leukemia treatment is not well known.
Sorafenibisa multi-targeting tyrosine kinase inhibitor with activity against B-RAF and other kinases such as FLT3, VEGF, PDGF receptor and c-KIT; and is used for the treatment of various malignancies including AML and ALL [55]. It was used by Feldmann et al., to investigated whether Sorafenib interferes with necroptosis in apoptosis-resistant acute leukemia. Feldmann reported that Sorafenib inhibits necroptotic signaling and cell death in two models of necroptosis in acute leukemia by significantly reducingsecond mitochondria-derived activator of caspases (Smac) mimetic-induced necroptosis in apoptosis-resistant Acute Myeloid Leukemia (AML) cells. Also, Smac mimetic/Tumor Necrosis Factor (TNF)α-inducenecroptosisin FADD-deficient Acute Lymphoblastic Leukemia (ALL) cells which implies that Sorafenib may limit the anti-leukemic activity of anticancer drugs that trigger necroptosis under certain conditions, for example when caspase activation is blocked [56].
Further, Ponatinib is a multikinase inhibitor, which targets BCR-ABL besides other kinases such as VEGF receptor, PDGF receptor, FGFR, FLT3 and c-KIT [57] and it has been shown to inhibit both RIP1 and RIP3 via direct binding [58]. Ponatinib has been developed for the treatment of Philadelphia chromosome-positive acute leukemia [59]. The protective effect of Pazopanib, a receptor tyrosine kinase inhibitor that targets VEGF receptors, PDGF receptor and c-KIT [60] has been reported to be mediated via RIP1 as the main functional target [58].Together, these data have shown that several multi-kinase inhibitors with different activity spectra can protect from necroptosis by targeting RIP1 and/or RIP3, therefore can limit the anti-leukemic activity of necroptosis-inducing drugs in acute leukemia cells which has important implications.
In AML, it had been recently demonstrated that Smacmimetics alone or in combination with standard chemotherapeutic drugs such as cytarabine or epigenetic modifiers (i.e. demethylating agents, Histone Deacetylase (HDAC) inhibitors) can overcome apoptosis resistance of AML cells by inducing necroptosis as an alternative mode of programmed cell death [61-64]. Also, Smacmimetics together with glucocorticoids, demethylating agents or TNFα have been shown to elicit necroptosis in apoptosis-resistant ALL cells [65-67].
Induction of cell death is an important strategy for killing cancer cells. Via the treatment of cancer, the cancerous cells develop resistance to apoptosis via defective caspase activity owing to gene mutations or silencing. Since the necroptosis tends to occur in the absence of caspase activation, it is conceivable that necroptosis is an alternative mode of cell death to overcome apoptosis resistance in leukemia cells. Therefore, triggering necroptosis may represent an attractive new strategy for apoptosis-resistance leukemia as well as cancer.
ACKNOWLEDGMENT
I express my sincere gratitude, and thanks to King Saud University (KSU) for providing the resources to complete my mini-review. I wish to express my sincere appreciation and deepest gratitude to the Deanship of Scientific Research and RSSU at King Saud University for their technical support.
REFERENCES
- Chaabane W, User SD, El-Gazzah M, Jaksik R, Sajjadi E, et al. (2013) Autophagy, apoptosis, mitoptosis and necrosis: Interdependence between those pathways and effects on cancer. Arch Immunol Ther Exp (Warsz) 61: 43-58.
- Edinger AL, Thomson CB (2004) Death by design: apoptosis, necrosis and autophagy. Curr Opin Cell Biol 16: 663-669.
- Kroemer G, Marino G, Levine B (2010) Autophagy and the integrated stress response. Mol Cell 40: 280-293.
- Curtin JF, Cotter TG (2003) Live and let die: regulatory mechanisms in Fas-mediated apoptosis. Cell Signal 15: 983-992.
- Los M, Wesselborg S, Schulze-osthoff K (1999) The role of caspases in development, immunity, and apoptotic signal transduction: lessons from knockout mice. Immunity 10: 629-639.
- Elmore S (2007) Apoptosis: a review of programmed cell death. Toxicol Pathol 35: 495-516.
- Ghavami S, Hashemi M, Ande SR, Yeganeh B, Xiao W, et al. (2009) Apoptosis and cancer: mutations within caspase genes. J Med Genet 46: 497-510.
- Suen DF, Norris KL, Youle RJ (2008) Mitochondrial dynamics and apoptosis. Genes Dev 22: 1577-1590.
- Chalah A, Khosravi-Far R (2008) The mitochondrial death pathway. Programmed Cell Death in Cancer Progression and Therapy 615: 25-45.
- Cory S, Adams JM (2005) Killing cancer cells by flipping the Bcl-2/Bax switch. Cancer Cell 8: 5-6.
- Rong Y, Distelhorst CW (2008) Bcl-2 Protein Family Members: Versatile Regulators of Calcium Signaling in Cell Survival and Apoptosis. Annu Rev Physiol 70: 73-91.
- Mizushima N (2007) Autophagy: process and function. Genes Dev 21: 2861-2873.
- Mizushima N, Levine B, Cuervo AM, Klionsky DJ (2008) Autophagy fights disease through cellular self-digestion. Nature 451: 1069-1075.
- Gump JM, Thorburn A (2011) Autophagy and apoptosis- what’s the connection? Trends Cell Biol 21: 387-392.
- Meijer AJ, Codogno P (2009) Autophagy: regulation and role in disease. Crit Rev Clin Lab Sci 46: 210-240.
- Isabel Colombo M, Simon HU (2009) Autophagy. Wiley online library.
- Mizushima N, Ohsumi Y, Yoshimori T (2002) Autophagosome formation in mammalian cells. Cell Struct Funct 27: 421-429.
- Eskelinen EL (2005) Maturation of autophagic vacuoles in mammalian cells. Autophagy 1: 1-10.
- Mizushima N, Noda T, Yoshimori T, Tanaka Y, Ishii T, et al. (1998) A protein conjugation system essential for autophagy. Nature 395: 395-398.
- Ichimura Y, Kirisako T, Takao T, Satomi Y, Shimonishi Y, et al. (2000) A ubiquitin-like system mediates protein lipidation. Nature 408: 488-492.
- Shintani T, Mizushima N, Ogawa Y, Matsuura A, Noda T, et al. (1999) Apg10p, a novel protein-conjugating enzyme essential for autophagy in yeast. EMBO J 18: 5234-5241.
- Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, et al. (2000) LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J 19: 5720-5728.
- Petiot A, Ogier-Denis E, Blommaart EF, Meijer AJ, Codogno P (2000) Distinct classes of phosphatidylinositol 3'-kinases are involved in signaling pathways that control macroautophagy in HT-29 cells. J Biol Chem 275: 992-998.
- Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, et al. (1999) Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 402: 672-676.
- Kabeya Y , Kamada Y, Baba M, Takikawa H, Sasaki M, et al. (2005) Atg17 functions in cooperation with Atg1 and Atg13 in yeast autophagy. Mol Biol Cell 16: 2544-2553.
- Kang R , Zeh HJ, Lotze MT, Tang D (2011) The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ 18: 571-580.
- Li BX, Li CY, Peng RQ, Wu XJ, Wang HY, et al. (2009) The expression of beclin 1 is associated with favorable prognosis in stage IIIB colon cancers. Autophagy 5: 303-306.
- Copetti T, Bertoli C, Dalla E, Demarchi F, Schneider C (2009) p65/RelA modulates BECN1 transcription and autophagy. Mol Cell Biol 29: 2594-2608.
- Tanida I, Ueno T, Kominami E (2008) LC3 and Autophagy. Methods Mol Biol 445: 77-88.
- Bell BD, Walsh CM (2009) Coordinate regulation of autophagy and apoptosis in T cells by death effectors: FADD or foundation. Autophagy 5: 238-240.
- Kinnally KW, Peixoto PM, Ryu SY, Dejean LM (2011) Is mPTP the gatekeeper for necrosis, apoptosis, or both? Biochim Biophys Acta 1813: 616-622.
- Smith CC, Yellon DM (2011) Necroptosis, necrostatins and tissue injury. J Cell Mol Med 15: 1797-1806.
- Chen W, Dang T, Blind RD, Wang Z, Cavasotto CN, et al. (2008) Glucocorticoid receptor phosphorylation differentially affects target gene expression. Mol Endocrinol 22: 1754-1766.
- Mason AR, Elia LP, Finkbeiner S (2017) The Receptor-interacting Serine/Threonine Protein Kinase 1 (RIPK1) Regulates Progranulin Levels. J Biol Chem 292: 3262-3272.
- Stanger BZ, Leder P, Lee TH, Kim E, Seed B )1995) RIP: a novel protein containing a death domain that interacts with Fas/APO-1 (CD95) in yeast and causes cell death. Cell 81: 513-523.
- Liu Y, Liu T, Lei T, Zhang D, Du S, et al. (2019) RIP1/RIP3-regulated necroptosis as a target for multifaceted disease therapy (Review). Int J Mol Med 44: 771-786.
- Christofferson DE, Yuan J (2010) Necroptosis as an alternative form of programmed cell death. Curr Opin Cell Biol 22: 263-268.
- Cho YS, Challa S, Moquin D, Genga R, Ray TD, et al. (2009) Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 137: 1112-1123.
- Holler N, Zaru R, Micheau O, Thome M, Attinger A, et al. (2000) Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat Immunol 1: 489-495.
- Vandenabeele P, Declercq W, Van Herreweghe F, VandenBerghe T (2010) The role of the kinases RIP1 and RIP3 in TNF-induced necrosis. Sci Signal 3: 4.
- Degterev A , Hitomi J, Germscheid M, Ch'en IL, Korkina O, et al. (2008) Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat Chem Biol 4: 313-321.
- Vanden Berghe T, Linkermann A, Jouan-Lanhouet S, Walczak H, Vandenabeele P (2014) Regulated necrosis: the expanding network of non-apoptotic cell death pathways. Nat Rev Mol Cell Biol 15: 135-147.
- He S, Wang L, Miao L, Wang T, Du F, et al. (2009) Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell 137: 1100-1111.
- Sun L, Wang H, Wang Z, He S, Chen S, et al. (2012) Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 148: 213-227.
- Wang H, Sun L, Su L, Rizo J, Liu L, et al. (2014) Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol Cell 54: 133-146.
- Dondelinger Y, Declercq W, Montessuit S, Roelandt R, Goncalves A, et al. (2014) MLKL Compromises Plasma Membrane Integrity by Binding to Phosphatidylinositol Phosphates. Cell Rep 7: 971-981.
- Fulda S (2014) Therapeutic exploitation of necroptosis for cancer therapy. Semin Cell Dev Biol 35: 51-56.
- Oberst A, Dillon CP, Weinlich R, McCormick LL, Fitzgerald P, et al. (2011) Catalytic activity of the caspase-8-FLIP(L) complex inhibits RIPK3-dependent necrosis. Nature 471: 363-367.
- Kroemer G, Martin SJ (2005) Caspase-independent cell death. Nat Med 11: 725-730.
- Reef S, Shifman O, Oren M, Kimchi A (2007) The autophagic inducer smARF interacts with and is stabilized by the mitochondrial p32 protein. Oncogene 26: 6677-6683.
- Djavaheri-Mergny M, Maiuri MC, Kroemer G (2010) Cross talk between apoptosis and autophagy by caspase-mediated cleavage of Beclin 1. Oncogene 29: 1717-1719.
- Luo S, Rubinsztein DC (2009) Apoptosis blocks Beclin 1-dependent autophagosome synthesis: an effect rescued by Bcl-xL. Cell Death Differ 17: 268-277.
- Maddika S, Ande SR, Panigrahi S, Paranjothy T, Weglarczyk K, et al. (2007) Cell survival, cell death and cell cycle pathways are interconnected: implications for cancer therapy. Drug Resist Updat 10: 13-29.
- Fulda S (2013) The mechanism of necroptosis in normal and cancer cells. Cancer Biol Ther 14: 999-1004.
- Daver N, Konopleva M (2015) Sorafenib and novel multikinase inhibitors in AML. Lancet Oncol 16: 1582-1583.
- Feldmann F, Schenk B, Martens S, Vandenabeele P, Fulda S (2017) Sorafenib inhibits therapeutic induction of necroptosis in acute leukemia cells. Oncotarget 8: 68208-68220.
- Miller GD, Bruno BJ, Lim CS (2014) Resistant mutations in CML and Ph(+)ALL - role of ponatinib. Biologics 8: 243-254.
- Fauster A, Rebsamen M, Huber KV, Bigenzahn JW, Stukalov A, et al. (2015) A cellular screen identifies ponatinib and pazopanib as inhibitors of necroptosis. Cell Death Dis 6: 1767.
- O’Hare T, Shakespeare WC, Zhu X, Eide CA, Rivera VM, et al. (2009) AP24534, a pan-BCR-ABL inhibitor for chronic myeloid leukemia, potently inhibits the T315I mutant and overcomes mutation-based resistance. Cancer Cell 16: 401-412.
- Harris PA, Boloor A, Cheung M, Kumar R, Crosby RM, et al. (2008) Discovery of 5-[[4-[(2,3-dimethyl-2H-indazol-6-yl)methylamino]-2-pyrimidinyl]amino]-2-methyl-benzenesulfonamide (Pazopanib), a novel and potent vascular endothelial growth factor receptor inhibitor. J Med Chem 51: 4632-4640.
- Safferthal C, Rohde K, Fulda S (2017) Therapeutic targeting of necroptosis by Smac mimetic bypasses apoptosis resistance in acute myeloid leukemia cells. Oncogene 36: 1487-1502.
- Steinwascher S, Nugues AL, Schoeneberger H, Fulda S (2015) Identification of a novel synergistic induction of cell death by Smac mimetic and HDAC inhibitors in acute myeloid leukemia cells. Cancer Lett 366: 32-43.
- Chromik J, Safferthal C, Serve H, Fulda S (2014) Smac mimetic primes apoptosis-resistant acute myeloid leukaemia cells for cytarabine-induced cell death by triggering necroptosis. Cancer Lett 344: 101-109.
- Steinhart L, Belz K, Fulda S (2013) Smac mimetic and demethylating agents synergistically trigger cell death in acute myeloid leukemia cells and overcome apoptosis resistance by inducing necroptosis. Cell Death Dis 4: 802.
- Rohde K, Kleinesudeik L, Roesler S, Lowe O, Heidler J, et al. (2017) A Bak-dependent mitochondrial amplification step contributes to Smac mimetic/glucocorticoid-induced necroptosis. Cell Death Differ 24: 83-97.
- Gerges S, Rohde K, Fulda S (2016) Cotreatment with Smac mimetics and demethylating agents induces both apoptotic and necroptotic cell death pathways in acute lymphoblastic leukemia cells. Cancer Lett 375: 127-132.
- Schenk B, Fulda S (2015) Reactive oxygen species regulate Smac mimetic/TNFα-induced necroptotic signaling and cell death. Oncogene 34: 5796-5806.
Citation: Qattan MYM (2019) (RIP1)/RIP3-Regulated Necroptosis, Interplay with Apoptosis and Autophagy, and Its Therapeutic Effect on Leukemia. J Stem Cell Res Dev Ther 5: 023.
Copyright: © 2019 Malak Yahia M Qattan, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.