Journal of Stem Cells Research Development & Therapy Category: Medical
Type: Research Article
Subcellular Localization of CD133 and Interleukin-6 Receptor (IL-6R) in Human Hepatoblastoma Cell-Line (HuH-6 Clone-5)
*Corresponding Author(s):
Masumi AkitaDivision Of Morphological Science, Biomedical Research Center, Saitama Medical University, 38 Moroyama, Iruma-gun, Saitama 350-0495, Japan
Tel:+81-49-276-2103,
Email:makita@saitama-med.ac.jp
Received Date: May 09, 2014
Accepted Date: Jun 17, 2014
Published Date: Jul 01, 2014
Abstract
We detected CD133 and interleukin-6 receptor (IL-6R) in the human hepatoblastoma cell-line (HuH-6 Clone-5). There was heterogeneity in the expression of CD133 and IL-6R at different stages. In the early stages of cultivation, CD133 and IL-6R were localized in the complex structure comprising filopodia and lamellipodia. In the late stages of cultivation, subcellular localization patterns of CD133 and IL-6R were different. CD133 was mainly localized to plasma membrane. After treatment with CD133 antibody, cell-cell contact was decreased, and apoptosis was increased. CD133-mediated regulation may be required for cell survival. IL-6R was mainly localized to dividing cells. Co-localization of CD133 and IL-6R were seen in some dividing cells. Present study suggests that co-localization of CD133 and IL-6R may play an important role of cell division.
Keywords
CD133; Plominin-1; IL-6R; Cancer stem cell; Asymmetric cell division; Hepatoblastoma; Immunohistochemistry; Cell culture; TEM; SEM
INTRODUCTION
CD133 (also known as Prominin-1 or AC133) has been identified as an important cell surface marker to enrich the stem-like population in various solid tumors, including colon [1,2], brain [3,4], skin [5], pancreatic [6], liver [7-10], prostate [11], and bone [12,13], tumors. CD133 was first isolated and cloned as an antigen present on the surface of mouse neuroepithelial progenitors [14], and human hematopoietic stem/progenitor cells [15,16]. CD133 expression is not restricted to the neuroepithelial and hematopoietic stem/progenitor cells, but extends to several epithelial and non-epithelial cell types [17]. However, the biological function of CD133 remains largely unknown. Hayashi et al. [10] separated CSCs by the side population (SP) fraction method from hepatoblastoma cells. They inoculated the SP fraction cells into NOD/SCID immunodeficient mice, and observed CD133 positive cells in the tumors formed in the mice. Tirino et al. [12] have shown that CD133+ cells from human bone sarcomas display high proliferation rate and that they are capable of forming cluster spheres (sarcospheres). Sarcospheres consisting CD133+ cells exhibited self-renewal, differentiation ability, tumorigenicity, and stemness gene expression [13].
Recently, we reported that CD133 was preferentially concentrated in a complex structure comprising filopodia and the leading edge of lamellipodia [18]. We also reported the co-localization of CD133 with F-actin. Furthermore, an antibody against CD133 decreased cell migration, strongly suggesting that CD133 is involved in tumor cell migration [18]. Co-localization of CD133 and IL-6Rwas reported in the lung cancer stem cells (CSCs) [19]. There is evidence that IL-6/6R signaling acts as a critical factor for the growth and malignancy of cancer cells [20,21]. The aberrant production and increased secretion of IL-6 in cancer patients is profoundly linked to tumor progression and poor prognosis in many cancer types [22-24]. However, the role of IL-6R in cancer stem cells (CSCs) is not well defined.IL-6R expression is up regulated in CSCs [25-27]. Yi et al. [19] reported that the expression of IL-6R in the lung CSCs was markedly up regulated at both the gene and protein levels. The prominent distribution of IL-6R was confirmed on the cell surface of CSCs, but not on non-CSCs. They also reported the co-localization of CD133 and IL-6R. However, they did not reveal the subcellular localization of CD133 and IL-6R.
In the present study, we detected CD133 and IL-6R in the human hepatoblastoma cell-line (HuH-6 Clone-5). We demonstrated that CD133 and IL-6R were preferentially concentrated in the complex structure comprising filopodia and lamellipodia.
In the late stages of cultivation, CD133 was mainly localized to the plasma membrane. IL-6R was mainly localized at the dividing cells. Some dividing cells expressed co-localization of CD133 and IL-6R. Co-localization of CD133 and IL-6R may play an important role of cell division for stemness.
Recently, we reported that CD133 was preferentially concentrated in a complex structure comprising filopodia and the leading edge of lamellipodia [18]. We also reported the co-localization of CD133 with F-actin. Furthermore, an antibody against CD133 decreased cell migration, strongly suggesting that CD133 is involved in tumor cell migration [18]. Co-localization of CD133 and IL-6Rwas reported in the lung cancer stem cells (CSCs) [19]. There is evidence that IL-6/6R signaling acts as a critical factor for the growth and malignancy of cancer cells [20,21]. The aberrant production and increased secretion of IL-6 in cancer patients is profoundly linked to tumor progression and poor prognosis in many cancer types [22-24]. However, the role of IL-6R in cancer stem cells (CSCs) is not well defined.IL-6R expression is up regulated in CSCs [25-27]. Yi et al. [19] reported that the expression of IL-6R in the lung CSCs was markedly up regulated at both the gene and protein levels. The prominent distribution of IL-6R was confirmed on the cell surface of CSCs, but not on non-CSCs. They also reported the co-localization of CD133 and IL-6R. However, they did not reveal the subcellular localization of CD133 and IL-6R.
In the present study, we detected CD133 and IL-6R in the human hepatoblastoma cell-line (HuH-6 Clone-5). We demonstrated that CD133 and IL-6R were preferentially concentrated in the complex structure comprising filopodia and lamellipodia.
In the late stages of cultivation, CD133 was mainly localized to the plasma membrane. IL-6R was mainly localized at the dividing cells. Some dividing cells expressed co-localization of CD133 and IL-6R. Co-localization of CD133 and IL-6R may play an important role of cell division for stemness.
MATERIALS AND METHODS
Cell culture
Human hepatoblastoma cells (HuH-6 clone 5, well-differentiated type, JCRB0401) were purchased from the Health Science Research Resources Bank (Osaka, Japan). According to a method previously reported by Hayashi et al. [10], the cells were cultured on plastic culture dishes (35 mm) or Aclar® plastic film (Nisshin EM, Tokyo, Japan) in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 50 µg/mL gentamicin. The cells were seeded at a density of 1 × 105 cells/ml in 35 mm culture dishes. Cultures were maintained at 37°C for 10 days in a humidified atmosphere of 5% CO2-95% air, and the medium was replaced every 2 days.
The Aclar® plastic films for electron microscopy were sterilized by immersion in 70% ethanol for 20 min. The films were then rinsed twice in PBS and placed on the bottom of plastic culture dishes (35 mm).
The Aclar® plastic films for electron microscopy were sterilized by immersion in 70% ethanol for 20 min. The films were then rinsed twice in PBS and placed on the bottom of plastic culture dishes (35 mm).
Immunohistochemistry for light microscopy
The cultured cells were fixed in 4% paraformaldehyde/PBS and streptavidin/peroxidase immunohistochemistry for CD133 and IL-6R was performed. The specimens were treated with 0.3% H2O2 in methanol to block endogenous peroxidase activity and subsequently incubated with primary antibodies CD133 (polyclonal antibody; Abnova, Taipei, Taiwan) and IL-6R (mouse monoclonal antibody; Santa Cruz Biotechnology, Inc., Santa Cruz, CA). A biotinylated, anti-rabbit immunoglobulin was added as a secondary antibody. A horseradish peroxidase-labelled streptomycin-avidin complex was then used to detect the secondary antibody. Antibody binding was detected by staining with the chromogens 3, 3'-diaminobenzidine (DAB).
Alexa Fluor 488-Fluoronanogold®-anti-rabbit Fab'(Nanoprobes, Inc., Yaphank, NY), Alexa Fluor 488-conjugated anti-rabbitFab' andAlexa Fluor 555-conjugated goat anti-mouse IgG (Molecular Probes, Inc., Eugene, OR) were also used for secondary antibodies. Diamidino-2-phenylindole (DAPI) staining was used to determine the number of nuclei.
Alexa Fluor 488-Fluoronanogold®-anti-rabbit Fab'(Nanoprobes, Inc., Yaphank, NY), Alexa Fluor 488-conjugated anti-rabbitFab' andAlexa Fluor 555-conjugated goat anti-mouse IgG (Molecular Probes, Inc., Eugene, OR) were also used for secondary antibodies. Diamidino-2-phenylindole (DAPI) staining was used to determine the number of nuclei.
Transmission Electron Microscopy (TEM)
The cultured cells were fixed in 0.1 M phosphate buffer (pH 7.2) containing 2.5% glutaraldehyde for 1 h, followed by fixation in 1% OsO4/0.1 M phosphate buffer (pH 7.2) for 1 hour. The specimens were dehydrated in a graded ethanol series, embedded in epoxy resin, cut into ultrathin sections, and stained with uranyl acetate and lead citrate. The stained ultrathin sections were observed under a transmission electron microscope (JEM-1400; Tokyo, Japan).
Scanning Electron Microscopy (SEM)
For SEM, the cultured cells were fixed in 2.5% glutaraldehyde/0.1 M phosphate buffer (pH 7.2) for 1 h and subsequently fixed in 1% OsO4/0.1 M phosphate buffer (pH 7.2) for 1 h, dehydrated in a graded ethanol series, and critical-point air-dried after treatment with isoamyl acetate. The samples were sputter coated with OsO4 and observed under a scanning electron microscope (Hitachi, S-4800; Tokyo, Japan).
Effect of CD133 antibody
According to a method previously reported by Akita et al. [18], cells were cultured to near confluence at 37°C in 35 mm culture dishes containing DMEM supplemented with 10% FBS. The cultures were treated with 2 concentrations of CD133 antibody (1 and 10 µg/mL treatment groups, 4 dishes per concentration) in fresh culture medium at 37°C in 5% CO2 for 48 h. One group is the control group and receives no treatment. After treatment with CD133 antibody, cells were treated for TEM, as described before. Ultrathin sections were observed under the transmission electron microscope (JEM-1400; Tokyo, Japan).
Statistical analysis
After treatment with CD133 antibody, number of cell divisions in the metaphase (the chromosomes align in the equator of the cell) and anaphase (the paired chromosomes separate and begin moving to opposite ends of the cell) was calculated. At least 10 photo images stained with DAPI were obtained from each dish. Each photo image approximately consists of 400 cells. Division index was expressed as a percentage of number of cell divisions/total cell number. Statistical differences were evaluated using the Student's t-test. P<0.01 was considered statistically significant. Data were expressed as mean ±SD values. Data represent the mean of triplicate experiments.
RESULTS
Localization of CD133 and IL-6R by light microscopy
Both CD133 positive and negative cells were observed. At the peripheral region of the cell, CD133 were strongly positive (Figure 1). Staining pattern of IL-6R was similar with those of the CD133. IL-6R was also strongly positive at the peripheral region of the cell (Figure 2). Electron microscopy revealed that CD133 and IL-6R were preferentially concentrated in a complex structure comprising filopodia and lamellipodia (Figures 3 and 4).
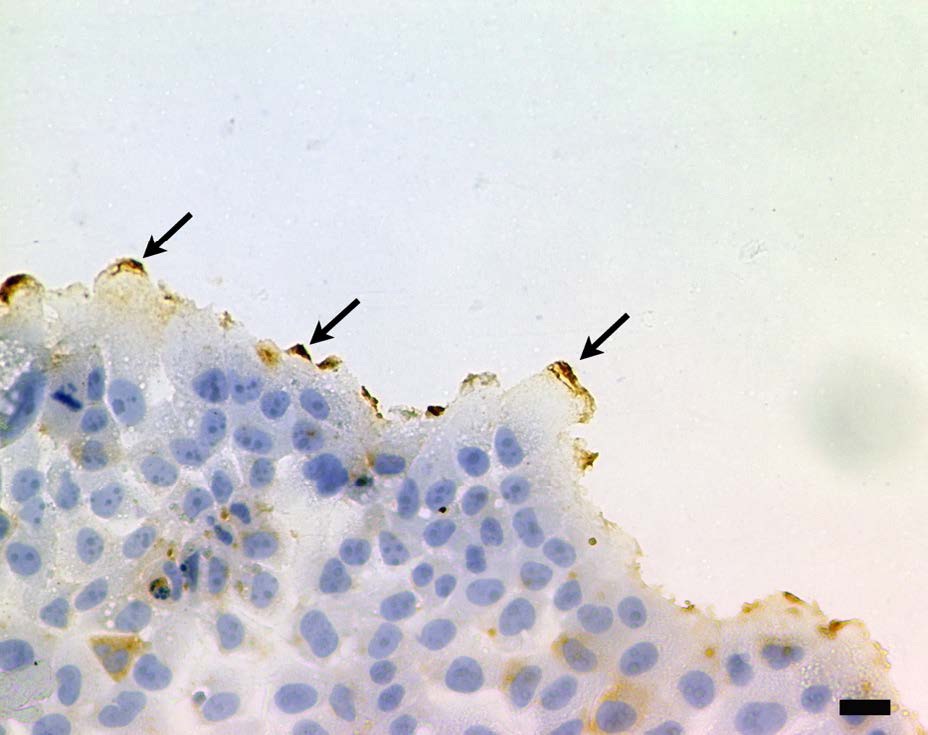 Figure 1: Localization of CD133 by light microscopy. Peripheralmembrane ruffles were strongly positive (arrows). DAB, brown precipitate and hematoxylin counterstain. Scale bar=20 μm.
Figure 1: Localization of CD133 by light microscopy. Peripheralmembrane ruffles were strongly positive (arrows). DAB, brown precipitate and hematoxylin counterstain. Scale bar=20 μm.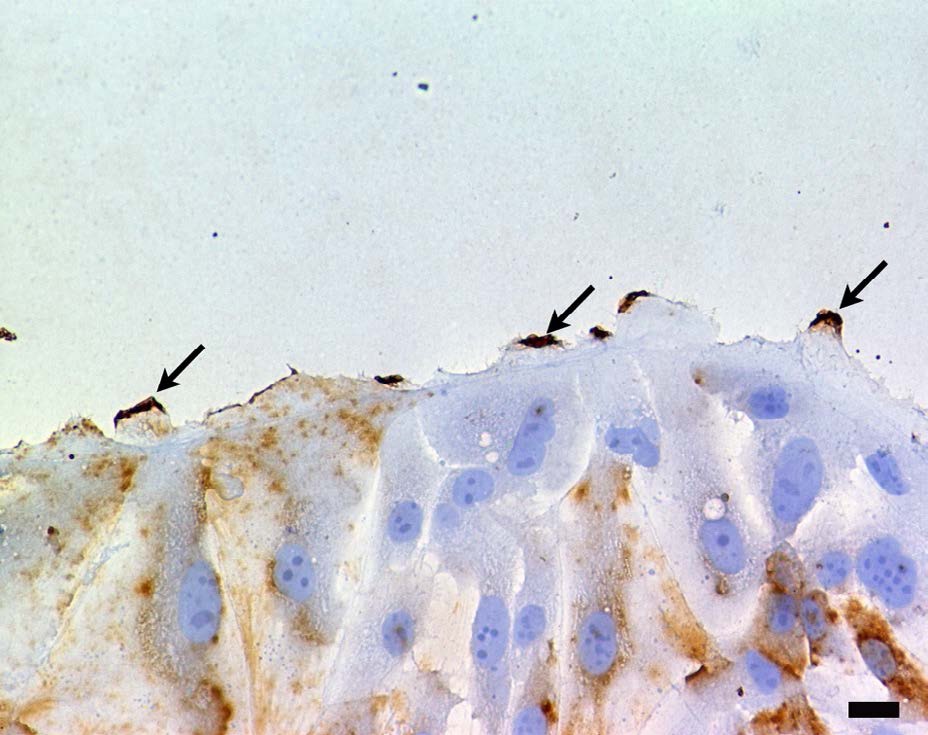 Figure 2: Localization of IL-6R by light microscopy. IL-6R was also positive at peripheral regions of the cell (arrows). DAB, brown precipitate and hematoxylin counter stain. Scale bar=20 μm.
Figure 2: Localization of IL-6R by light microscopy. IL-6R was also positive at peripheral regions of the cell (arrows). DAB, brown precipitate and hematoxylin counter stain. Scale bar=20 μm.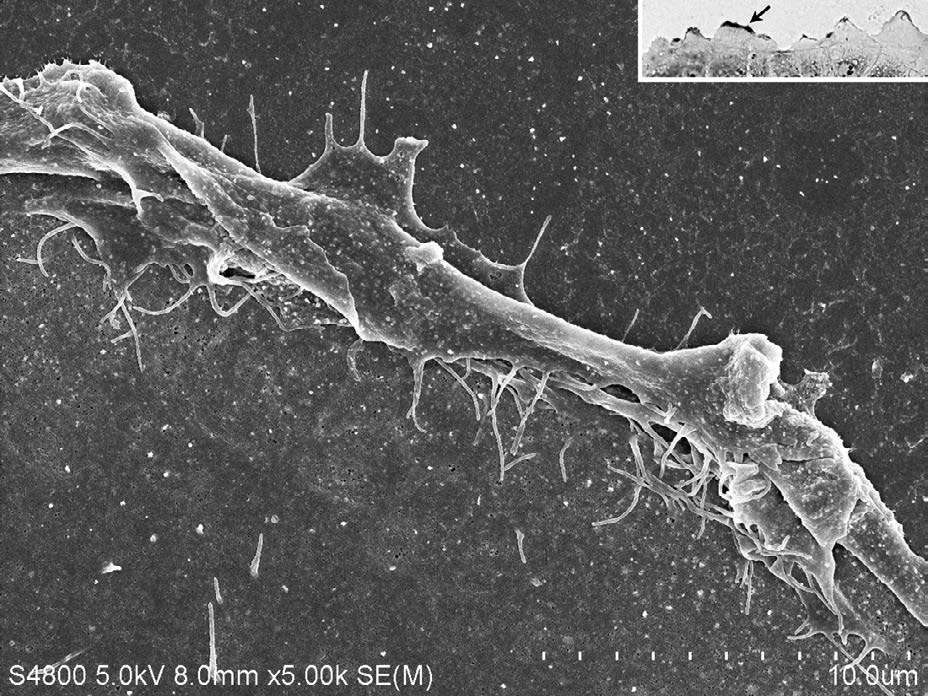
Figure 3: CD133 positive site indicated by the arrow (inset) was observed by SEM. Enlarged SEM image of filopodia and lamellipodia. Complex structure of filopodia and lamellipodia was coincident with the positive site.
Inset; Light microscopy; CD133 was strongly positive at the peripheral region of cell (arrow). Alexa Fluor 488-and Fluoronanogold (1.4 nm). Antibody binding site was enhanced using Gold Enhance at room temperature for 20.
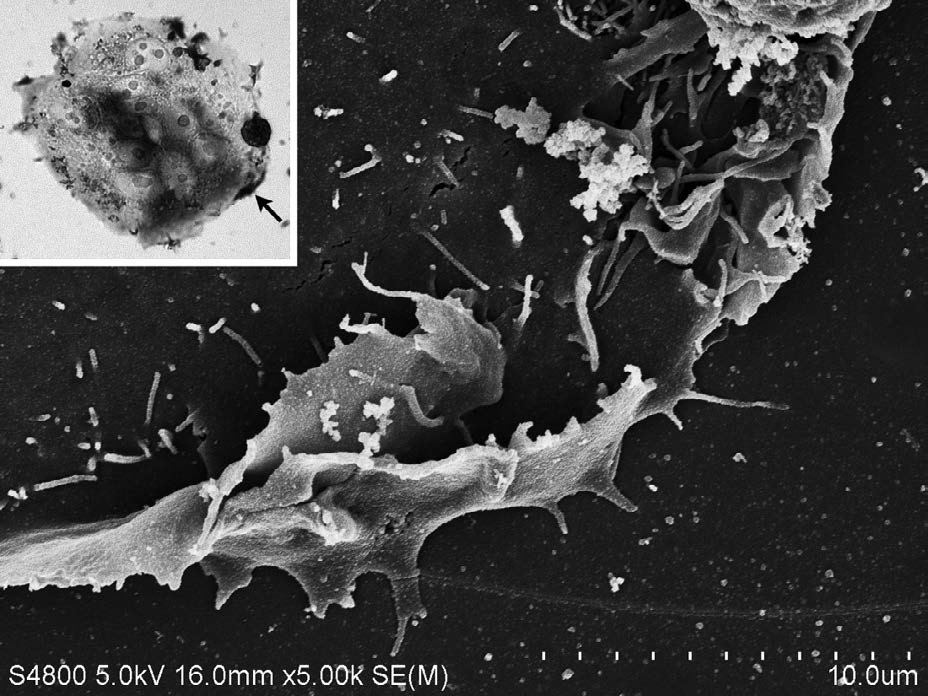
Figure 4: Enlarged SEM image of CD133 positive site indicated by the arrow (inset). CD133 positive site was observed as a complex structure of filopodia and lamellipodia.
Inset; Phase contrast microscopy; IL-6R was strongly positive at the peripheral region of cell (arrow). Antibody binding site was detected by the staining with DAB.
Time-dependent localization of CD133 and IL-6R
Culture of early stages (<2-day-old)
Co-localization of CD133 andIL6-R was partially observed. However, the expression of CD133 and IL-6R was heterogeneous (Figure 5).
Co-localization of CD133 andIL6-R was partially observed. However, the expression of CD133 and IL-6R was heterogeneous (Figure 5).
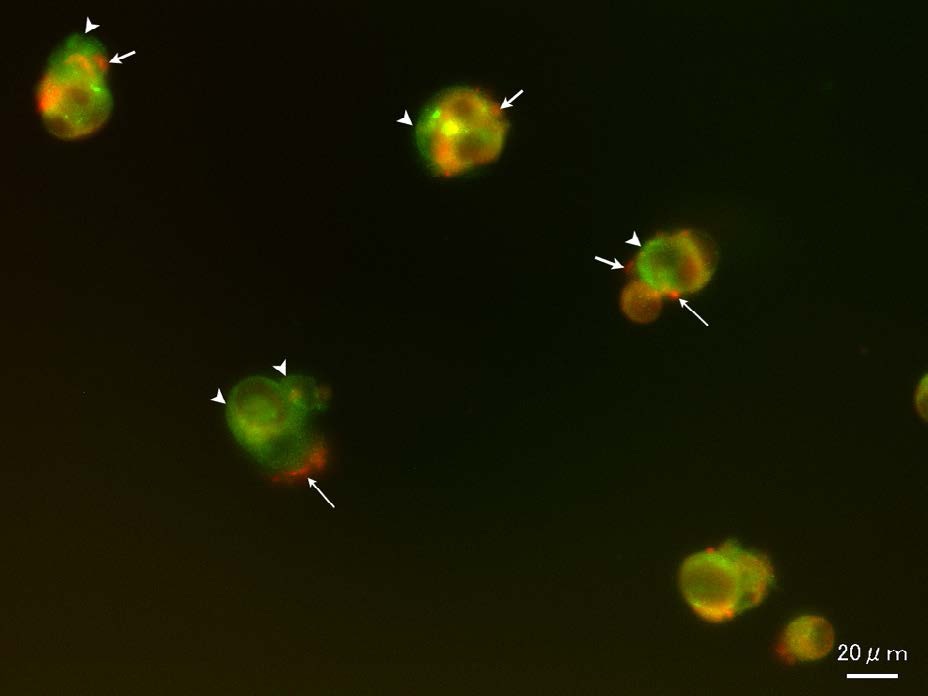 Figure 5: Culture of the early stage (2-day-old). Localization of CD133 and IL-6R. Co-localization of CD133 andIL6-R was partially observed. However, the expression of CD133 and IL-6R was heterogeneous. CD133 positive sites were labeled in green (arrowheads), IL-6R in red (arrows). CD133 and IL-6R positive sites were labeled in orange.
Figure 5: Culture of the early stage (2-day-old). Localization of CD133 and IL-6R. Co-localization of CD133 andIL6-R was partially observed. However, the expression of CD133 and IL-6R was heterogeneous. CD133 positive sites were labeled in green (arrowheads), IL-6R in red (arrows). CD133 and IL-6R positive sites were labeled in orange.Culture of late stages (7-day-old<)
CD133 was mainly localized to the plasma membrane. IL-6R was mainly localized at the dividing cells (Figure 6). Unequal distribution of CD133 was observed in some cells (Figure 7).
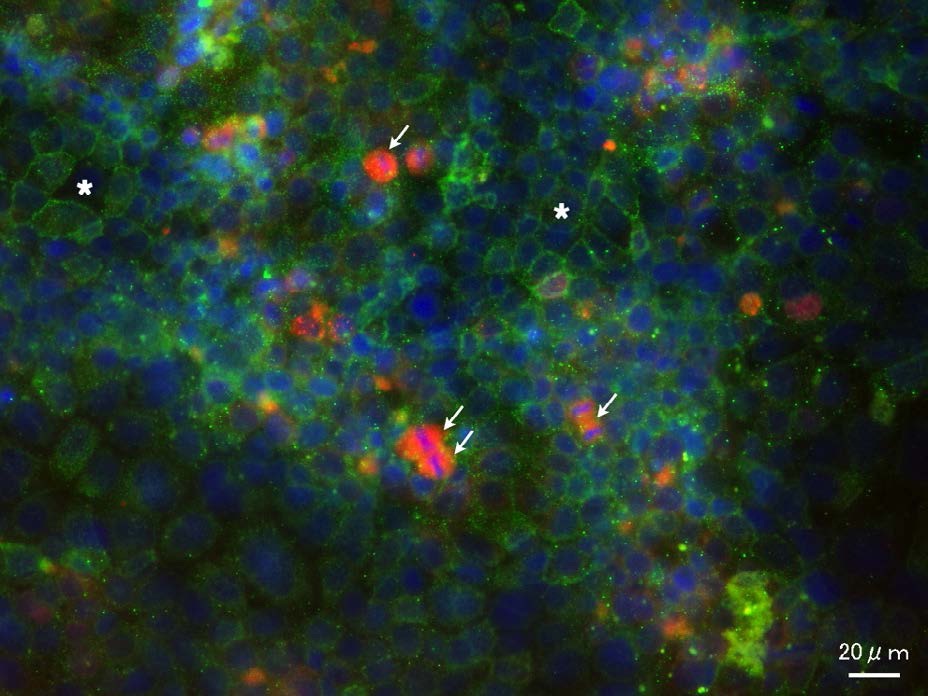
Figure 6: Culture of the late stage (10-day-old). Localization of CD133 and IL-6R.
CD133 was mainly localized to the plasma membrane. CD133 positive sites were observed as a mesh-like structure (asterisks). IL-6R was mainly localized to some dividing cells (arrows).
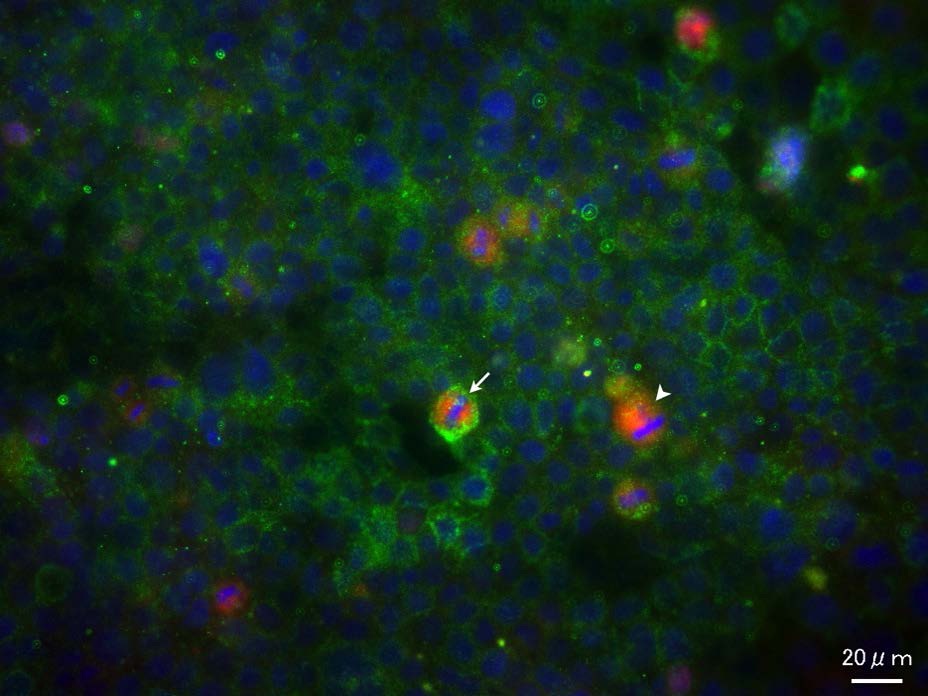 Figure 7: Co-localization of CD133 and IL-6R.Unequal distribution of CD133 was observed in some cells (arrow). IL-6R positive cell was indicated by arrowhead.
Figure 7: Co-localization of CD133 and IL-6R.Unequal distribution of CD133 was observed in some cells (arrow). IL-6R positive cell was indicated by arrowhead.Effect of CD133 antibody
After treatment with CD133 antibody, number of cell divisions in the metaphase and anaphase was calculated. Treatment with CD133 antibody (10 µg/mL) markedly decreased the number of cell divisions. (Figure 8) showed the division index.
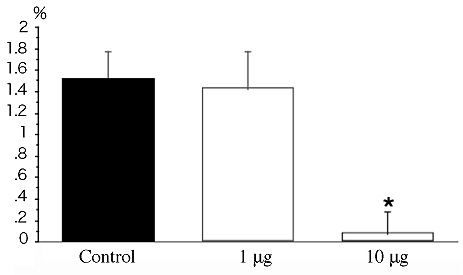
Figure 8: Effect of CD133 antibody treatment. After treatment with CD133 antibody (1 and 10 μg/mL), number of cell divisions in the metaphase and anaphase was calculated.
Bar graphs showing the division index (number of cell divisions/total cell number).
There was significant difference (*P<0.01) between CD133 antibody treatment (10 μg/mL) and control. However, the division index was not significantly affected at the lower antibody concentration (1 μg/mL).
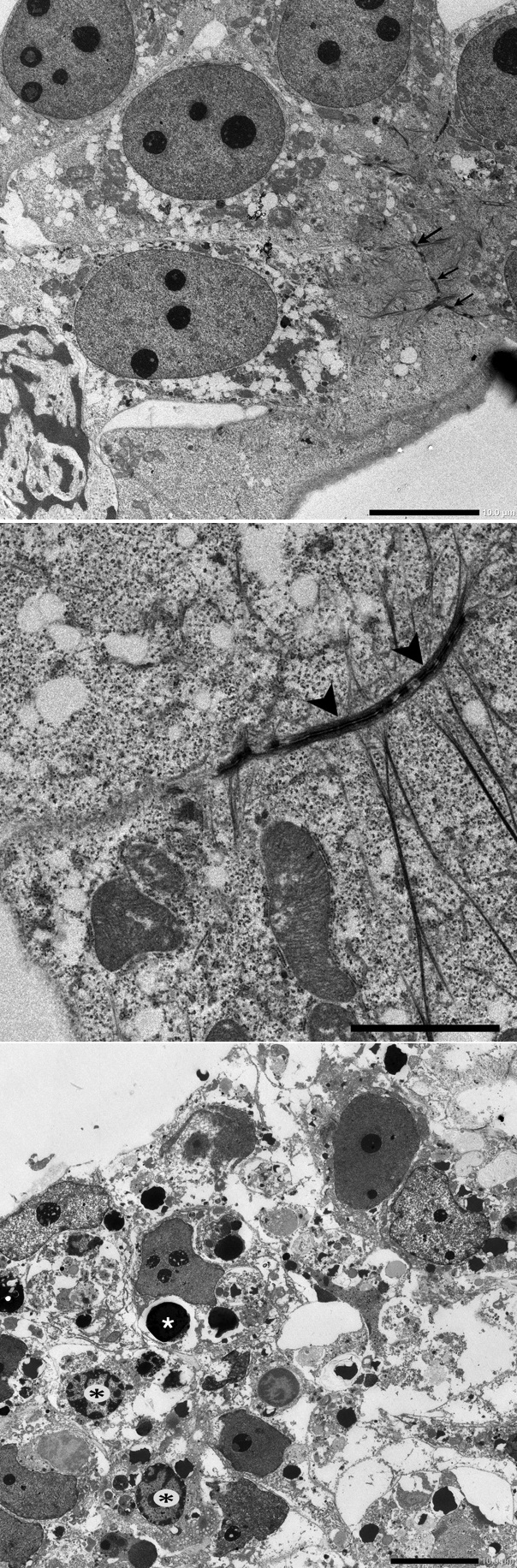
Figure 9: Morphological influence of CD133 anti body treatment.
A: Control; Nuclei and nucleoli were clearly observed. Cell-cell adhesion (arrows) was well defined. Scale bar=10 μm.
B: Control; Arrowheads indicate desmosomal structure. Scale bar=5 μm.
C: CD133 antibody (10 μg/mL); Apoptotic cells (black asterisks) and apoptotic body (white asterisk) were observed. Cell-cell adhesion was diminished, and desmosomal structure was not observed. Scale bar=10 μm.
DISCUSSION
CD133 is concentrated in microspikes, filopodia and the leading edge of lamellipodia in the fibroblasts expressing transfected CD133 [28]. Previously, we revealed that CD133 was preferentially concentrated in a complex structure comprising filopodia and lamellipodia [18]. In the present study, IL-6R was also concentrated at the similar site. It is assumed that CD133 and IL-6R relate to cell migration.
In the present study, we also demonstrated that the expression of CD133 and IL-6R was heterogeneous in different stages. In the late stages of cultivation, CD133 and IL-6R exhibited different patterns of subcellular localization. CD133 was mainly localized to the plasma membrane. Koyama-Nasu et al. [29] have shown that CD133 interacts with plakoglobin (also known as c-catenin), a desmosomal linker protein. They further demonstrate that knockdown of CD133 by RNA interference (RNAi) results in the down regulation of desmoglein-2, a desmosomal cadherin, and abrogate cell-cell adhesion and tumorigenicity of clear cell carcinoma of the ovary stem cells. In the present study, we confirmed the diminishing cell adhesion by decreasing desmosomal structure, after treatment with CD133 antibody. Present result obtained from CD133 antibody treatment also suggests that CD133-mediated regulation may be required for cell survival.
Co-localization of CD133 and IL-6R was demonstrated in the dividing cells. Some cells indicated an unequal distribution of CD133. Normal tissue homeostasis is maintained through asymmetric cell divisions that produce daughter cells with differing self-renewal and differentiation potentials [30]. Certain tumor cell subfractions can self-renew and repopulate the heterogeneous tumor bulk, suggestive of asymmetric cell division, but an equally plausible explanation is that daughter cells of a symmetric division subsequently adopt differing cell fates [30]. Asymmetric cell division is a proposed mechanism to maintain CSCs. Lathia et al. [31] utilized glioma stem cells (GSCs) for two cell division modes. Through a symmetric cell division, two GSCs were generated. The other mode is to generate one GSC and one non-GSC through an asymmetric cell division. This division mode would contribute to increased cellular heterogeneity of the tumor, while still maintaining a GSC pool [31]. Co-localization of CD133 and IL-6R may play an important role of cell division. Further studies are required to understand the relationship between symmetric cell division and CSCs.
IL-6R has been targeted for cancer treatments, resulting in cell growth inhibition or reduced angiogenesis [32-34]. Wang et al. [27] showed that knockdown of IL-6R diminished stemness and survival of glioma stem cells. Present study suggests that CD133 and IL-6R are targeted for cancer treatments, resulting in inhibition of cell proliferation and increasing apoptosis.
In the present study, we also demonstrated that the expression of CD133 and IL-6R was heterogeneous in different stages. In the late stages of cultivation, CD133 and IL-6R exhibited different patterns of subcellular localization. CD133 was mainly localized to the plasma membrane. Koyama-Nasu et al. [29] have shown that CD133 interacts with plakoglobin (also known as c-catenin), a desmosomal linker protein. They further demonstrate that knockdown of CD133 by RNA interference (RNAi) results in the down regulation of desmoglein-2, a desmosomal cadherin, and abrogate cell-cell adhesion and tumorigenicity of clear cell carcinoma of the ovary stem cells. In the present study, we confirmed the diminishing cell adhesion by decreasing desmosomal structure, after treatment with CD133 antibody. Present result obtained from CD133 antibody treatment also suggests that CD133-mediated regulation may be required for cell survival.
Co-localization of CD133 and IL-6R was demonstrated in the dividing cells. Some cells indicated an unequal distribution of CD133. Normal tissue homeostasis is maintained through asymmetric cell divisions that produce daughter cells with differing self-renewal and differentiation potentials [30]. Certain tumor cell subfractions can self-renew and repopulate the heterogeneous tumor bulk, suggestive of asymmetric cell division, but an equally plausible explanation is that daughter cells of a symmetric division subsequently adopt differing cell fates [30]. Asymmetric cell division is a proposed mechanism to maintain CSCs. Lathia et al. [31] utilized glioma stem cells (GSCs) for two cell division modes. Through a symmetric cell division, two GSCs were generated. The other mode is to generate one GSC and one non-GSC through an asymmetric cell division. This division mode would contribute to increased cellular heterogeneity of the tumor, while still maintaining a GSC pool [31]. Co-localization of CD133 and IL-6R may play an important role of cell division. Further studies are required to understand the relationship between symmetric cell division and CSCs.
IL-6R has been targeted for cancer treatments, resulting in cell growth inhibition or reduced angiogenesis [32-34]. Wang et al. [27] showed that knockdown of IL-6R diminished stemness and survival of glioma stem cells. Present study suggests that CD133 and IL-6R are targeted for cancer treatments, resulting in inhibition of cell proliferation and increasing apoptosis.
ACKNOWLEDGMENT
This work was partly supported by the programs of the Grant-in-Aid for Scientific Research (C) from the Japan Society for the Promotion of Science (JSPS) KAKENHI, Grant Number 25462779.
CONFLICT OF INTEREST
The author declares no competing financial interests.
REFERENCES
- O'Brien CA, Pollett A, Gallinger S, Dick JE (2007) A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature 445: 106-110.
- Ricci-Vitiani L, Lombardi DG, Pilozzi E, Biffoni M, Todaro M, et al. (2007) Identification and expansion of human colon-cancer-initiating cells. Nature 445: 111-115.
- Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, et al. (2004) Identification of human brain tumour initiating cells. Nature 432: 396-401.
- Liu G, Yuan X, Zeng Z, Tunici P, Ng H, et al. (2006) Analysis of gene expression and chemoresistance of CD133+ cancer stem cells in glioblastoma. Mol Cancer 5: 67.
- Monzani E, Facchetti F, Galmozzi E, Corsini E, Benetti A, et al. (2007) Melanoma contains CD133 and ABCG2 positive cells with enhanced tumourigenic potential. Eur J Cancer 43: 935-946.
- Olempska M, Eisenach PA, Ammerpohl O, Ungefroren H, Fandrich F, et al. (2007) Detection of tumor stem cell markers in pancreatic carcinoma cell lines. Hepatobiliary Pancreat Dis Int 6: 92-97.
- Suetsugu A, Nagaki M, Aoki H, Motohashi T, Kunisada T, et al. (2006) Characterization of CD133+ hepatocellular carcinoma cells as cancer stem/progenitor cells. Biochem Biophys Res Commun 351: 820-824.
- Ma S, Chan KW, Hu L, Lee TK, Wo JY, et al. (2007) Identification and characterization of tumorigenic liver cancer stem/progenitor cells. Gastroenterology 132: 2542-2556.
- Yin S, Li J, Hu C, Chen X, Yao M, et al. (2007) CD133 positive hepatocellular carcinoma cells possess high capacity for tumorigenicity. Int J Cancer 120: 1444-1450.
- Hayashi S, Fujita K, Matsumoto S, Akita M, Satomi A (2011) Isolation and identification of cancer stem cells from a side population of a human hepatoblastoma cell line, HuH-6 clone-5. Pediatr Surg Int 27: 9-16.
- Collins AT, Berry PA, Hyde C, Stower MJ, Maitland NJ (2005) Prospective identification of tumorigenic prostate cancer stem cells. Cancer Res 65: 10946-10951.
- Tirino V, Desiderio V, d'Aquino R, De Francesco F, Pirozzi G, et al. (2008) Detection and characterization of CD133+ cancer stem cells in human solid tumours. PLoS One 3: e3469.
- Tirino V, Desiderio V, Paino F, De Rosa A, Papaccio F, et al. (2011) Human primary bone sarcomas contain CD133+ cancer stem cells displaying high tumorigenicity in vivo. FASEB J 25: 2022-2030.
- Weigmann A, Corbeil D, Hellwig A, Huttner WB (1997) Prominin, A novel microvilli specific polytopic membrane protein of the apical surface of epithelial cells, is targeted to plasmalemmal protrusions of nonepithelial cells. Proc Natl Acad Sci USA 94: 12425-12430.
- Yin AH, Miraglia S, Zanjani ED, Almeida-Porada G, Ogawa M, et al. (1997) AC133, a novel marker for human hematopoietic stem and progenitor cells. Blood 90: 5002-5012.
- Miraglia S, Godfrey W, Yin AH, Atkins K, Warnke R, et al. (1997) A novel five- transmembrane hematopoietic stem cell antigen: isolation, characterization, and molecular cloning. Blood 90: 5013-5021.
- Corbeil D, Röper K, Fargeas CA, Joester A, Huttner WB (2001) Prominin: A Story of Cholesterol, Plasma Membrane Protrusions and Human Pathology. Traffic 2: 82-91.
- Akita M, Tanaka K, Murai N, Matsumoto S, Fujita K, et al. (2013) Detection of CD133 (prominin-1) in a human hepatoblastoma cell line (HuH-6 clone 5). Microsc Res Tech 76: 844-852.
- Yi H, Cho HJ, Cho SM, Jo K, Park JA, et al. (2012) Blockade of interleukin-6 receptor suppresses the proliferation of H460 lung cancer stem cells. Int J Oncol 41: 310-316.
- Hirano T, Ishihara K, Hibi M (2000) Roles of STAT3 in mediating the cell growth, differentiation and survival signals relayed through the IL-6 family of cytokine receptors. Oncogene 19: 2548-2556.
- Jee SH, Chu CY, Chiu HC, Huang YL, Tsai WL, et al. (2004) Interleukin-6 induced basic fibroblast growth factor-dependent angiogenesis in basal cell carcinoma cell line via JAK/STAT3 and PI3-kinase/Akt pathways. J Invest Dermatol 123: 1169-1175.
- Hodge DR, Hurt EM, Farrar WL (2005) The role of IL-6 and STAT3 in inflammation and cancer. Eur J Cancer 41: 2502-2512.
- Tchirkov A, Khalil T, Chautard E, Mokhtari K, Véronèse L, et al. (2007) Interleukin-6 gene amplification and shortened survival in glioblastoma patients. Br J Cancer 96: 474-476.
- Gao SP, Mark KG, Leslie K, Pao W, Motoi N, et al. (2007) Mutations in the EGFR kinase domain mediate STAT3 activation via IL-6 production in human lung adenocarcinomas. J Clin Invest 117: 3846-3856.
- Seo DC, Sung JM, Cho HJ, Yi H, Seo KH, et al. (2007) Gene expression profiling of cancer stem cell in human lung adenocarcinoma A549 cells. Mol Cancer 6: 75.
- Levina V, Marrangoni AM, DeMarco R, Gorelik E, Lokshin AE (2008) Drug-selected human lung cancer stem cells: cytokine network, tumorigenic and metastatic properties. PLoS One 3:e3077.
- Wang H, Lathia JD, Wu Q, Wang J, Li Z, et al. (2009) Targeting interleukin 6 signaling suppresses glioma stem cell survival and tumor growth. Stem Cells 27: 2393-2404.
- Corbeil D, Röper K, Hellwig A, Tavian M, Miraglia S, et al. (2000) The human AC133 hematopoietic stem cell antigen is also expressed in epithelial cells and targeted to plasma membrane protrusions. J Biol Chem 275: 5512-5520.
- Koyama-Nasu R, Takahashi R, Yanagida S, Nasu-Nishimura Y, Oyama M, et al. (2013) The cancer stem cell marker CD133 interacts with plakoglobin and controls desmoglein-2 protein levels. PLoS One 8: e53710.
- Pine SR, Ryan BM, Varticovski L, Robles AI, Harris CC (2010) Microenvironmental modulation of asymmetric cell division in human lung cancer cells. PNAS 107: 2195-2200.
- Lathia JD, Hitomi M, Gallagher J, Gadani SP, Adkins J, et al. (2011) Distribution of CD133 reveals glioma stem cells self-renew through symmetric and asymmetric cell divisions. Cell Death and Disease 2: e200.
- Kudo M, Jono H, Shinriki S, Yano S, Nakamura H, et al. (2009) Antitumor effect of humanized anti-interleukin-6 receptor antibody (tocilizumab) on glioma cell proliferation. Laboratory investigation. J Neurosurg 111: 219-225.
- Shinriki S, Jono H, Ota K, Ueda M, Kudo M, et al. (2009) Humanized anti-interleukin-6 receptor antibody suppresses tumor angiogenesis and in vivo growth of human oral squamous cell carcinoma. Clin Cancer Res 15: 5426-5434.
- Jones SA, Scheller J, Rose-John S (2011) Therapeutic strategies for the clinical blockade of IL-6/gp130 signaling. J Clin Invest 121: 3375-3383.
Citation: Akita M, Matsumoto S, Murai N, Komatsu K, Fujita K (2014) Subcellular Localization of CD133 and Interleukin-6 Receptor (IL-6R) in Human Hepatoblastoma Cell-Line (HuH-6 Clone-5). J Stem Cell Res Dev Ther, 1: 001.
Copyright: © 2014 Masumi Akita, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Journal Highlights
© 2026, Copyrights Herald Scholarly Open Access. All Rights Reserved!