The Role of Iron Homeostasis in Alzheimer’s Disease
*Corresponding Author(s):
Manolov VictorDepartment Of Clinical Laboratory And Clinical Immunology, Medical University, Sofia, Bulgaria
Tel:+359 29230928,
Fax:+359 29230922
Email:victhedoc2@yahoo.com
Abstract
Background
Alzheimer’s Disease (AD) is characterized by deposition of amyloid plaques of amyloid-β chelating peptide with transition metal ions (Cu2+, Zn2+ and Fe3+). The binding of Cu2+ and Fe3+ leads to toxic chemical reactions; a change in the oxidation of two metals, that leads to H2O2 production in the presence of transition metals and finally gives toxic free OH• radicals.
Methods
58 Alzheimer’s disease patients were included in this study. They were evaluated for serum iron, copper, selenium, zinc and hepcidin levels. Superoxide Dismutase (SOD) and Glutathione Peroxidase (GPX) were measured as oxidative stress markers. Hepcidin, SOD and GPX were measured by ELISA methods. Serum Fe, Cu, Se and Zn were quantified by AAS. The results form AD patients were compared to age and gender matched healthy controls. We used Pearson’s correlation and Student’s paired t-test for statistical analysis of established results.
Results
We found statistically significant elevated serum iron, copper and zinc results in AD patients (41.4 µmol/l, 37.8 µmol/l, and 39.9 µmol/l) compared to control group (20.6 µmol/l, 19.1 µmol/l, and 14.8 µmol/l); P<0.01. Plasma selenium levels were decreased in AD patients (114.8 nmol/L) compared to healthy controls (629.6 nmol/L); P<0.005. Hepcidin concentrations were increased in AD cases (64.9 µg/l) compared to controls (22.3 µg/l); P<0.001. SOD and GPX levels were decreased in Alzheimer’s disease (8.1 µg/ml, and 10.5 pg/mL) compared to normal values in healthy controls (20.4 µg/ml, and 37.4 pg/mL); P<0.001.
Conclusions
The expected contribution from our study is practical introduction of quantification of serum hepcidin as a potential marker for early diagnosis of impaired iron homeostasis, leading trace element in the pathogenesis of neurodegenerative diseases.
Keywords
ABBREVIATIONS
AAS: Atomic Absorption Spectrometry
AD: Alzheimer’s Disease
ALS: Amyotrophic Lateral Sclerosis
Aβ: Amyloid-β
BBB: Blood-Brain Barrier
Cu: Copper
CBC: Complete Blood Count
CHr: Concentration of Hemoglobin in Reticulocytes
DMT1: Divalent Metal Transporter 1
ELISA: Enzyme Linked Immunosorbent Assay
Fe: Iron
GPX: Glutathione Peroxidase
HD: Huntington’s Disease
HIF-1: Hypoxia Inducible Factor 1
HIF2α: Hypoxia Inducible Factor 2α
PD: Parkinson’s Disease
RNS: Reactive Nitrogen Species
ROS: Reactive Oxygen Species
Se: Selenium
SOD: Superoxide Dismutase
TIBC: Total Iron Binding Capacity
Zn: Zinc
INTRODUCTION
Oxygen is needed for energy metabolism and normal function of most eukaryotic organisms. It is partially reduced in the respiratory chain of low superoxide free basic radical, which can be converted into other forms of Reactive Oxygen Species (ROS). Cellular metabolism could generate other free radicals from nitrogen classified in the family of Reactive Nitrogen Species (RNS). ROS and RNS in physiological concentrations mediates a variety of normal functions, such as signal transduction regulation, induction of mitogenic response, even moderates the defense against infectious agents and others.
ROS are balanced with antioxidant systems to maintain a constant level in the body. These antioxidant systems are enzymatic and non-enzymatic. Violation of the balance of overproduction of ROS and/or reduction of antioxidants is called oxidative stress. Under these conditions, the large amount of free radicals may pass through the plasma membrane, damages the cell membrane through lipid peroxidation, and modifies the signal and structural proteins, resulting in oxidation of the RNA/DNA, transcription termination, and occurrence of gene mutation.
The clearest aging effect in neuronal degeneration usually is associated with impaired trace elements metabolism -Cu, Fe and Zn. The aging brain becomes more susceptible to degenerative processes [1].
Neurodegenerative diseases are conditions in which the nervous system progressively and irreversibly deteriorates. Neurodegenerative diseases are often with late manifestation of disorders, including Alzheimer’s Disease (AD), Parkinson’s Disease (PD), Huntington’s Disease (HD) and Amyotrophic Lateral Sclerosis (ALS).
AD depends on age; its a chronic neurodegenerative disease, which leads to dementia among the elderly. It is well known that AD is characterized by amyloid plaques deposition with transition metal ions (Cu2+, Zn2+and Fe3+) [2].
Indirect mechanisms exist by which Hypoxia-Inducible Factor 1 (HIF-1) regulates hepcidin expression. Increased HIF activity is connected to secession of hemojuvelin from matriptase-2, which down regulates hepcidin expression [3]. Systematic iron regulation from hepcidin-ferroportin axis collaborates with control mechanisms on enterocytes cellular level. In mouse models deficiency of duodenal Hypoxia-Inducible Factor 2α (HIF2α) is observed decreased expression not only for ferroportin, but for Divalent Metal Transporter 1 (DMT1), which is the main iron exporter and is situated on apical surface of duodenal enterocytes. It is unable to place absorption, even if hepcidin secretion is decreased [4]. This might bring to conclusion, that in case of hypoxia, HIF2α expression leads to iron absorption from intestinal bowels through increased DMT1 and ferroportin activity. Therefore HIF2α is able to change iron transport, despite of hepcidin-ferroportin axis regulation.
Oxidative stress plays an important role in neuronal damage caused by cerebral ischemia. It was found that free iron is significantly increased during ischemia and is responsible for the oxidative damage of the brain. A number of mechanisms are involved in brain injury by oxidative stress [5,6]. Iron plays a critical role in neuronal damage caused by oxidative stress. Increased levels of Hypoxia Inducible Factor 1 (HIF-1) is an expression of high hepcidin secretion [7,8].
METHODS
58 patients with Alzheimer’s disease were included. Their results were compared to age and gender matched healthy controls. Alzheimer’s disease patients were monitored in “Aleksandrovska” hospital, Clinic of Neurology.
Atomic Absorption Spectrometry (AAS- Perkin Elmer) was used for quantification of serum iron and Total Iron Binding Capacity (TIBC), serum Cu and Zn, and plasma Se. Serum ferritin levels were quantified by CLIA method (Immulyte 2000, by Siemens Healthcare). CBC, including hemoglobin concentration in reticulocytes (CHr) was quantified on ADVIA 2120 (provided by Siemens Healthcare). All chemical laboratory parameters were specified on Dimension RxL MAX (Siemens Healthcare Diagnostics). Included groups were evaluated for serum iron, copper, selenium, zinc and hepcidin levels. Superoxide Dismutase (SOD) and Glutathione Peroxidase (GPX) were measured as oxidative stress markers. Hepcidin, SOD and GPX were measured by ELISA methods (on VersaMax ELISA reader). Paired Student’s t-test and Pearson’s correlation were used for statistical analysis. Enrolled patients signed written informed consent according to the Declaration of Helsinki (Directive 2001/20/ EC).
RESULTS
Age distribution of patients in the different included groups is shown in table 1.
| Group | Male | Age |
| Alzheimer’s disease | 28 (48.3%) | 74.1 ± 8.6 |
| Control group | 28 (48.3%) | 73.9 ± 7.1 |
Table 1: Age distribution of patients in included groups.
Established parameters from CBC and laboratory assessment are presented in table 2.
| Parameter | AD | Control group | ||||
| Mean | Min | Max | Mean | Min | Max | |
| Hb (g/L) | 139.5 | 118.0 | 161.0 | 141.8 | 121.9 | 161.7 |
| RBC (*1012/L) | 4.01 | 3.21 | 4.81 | 4.75 | 4.05 | 5.45 |
| CHr (pg) | 23.7 | 21.1 | 26.3 | 31.9 | 29.1 | 34.7 |
| Creatinine (µmol/L) | 70.9 | 65.8 | 76.0 | 75.6 | 70.3 | 80.9 |
| Iron (µmol/L) | 41.4 | 37.9 | 44.9 | 20.6 | 18.4 | 22.8 |
| TIBC (µmol/L) | 75.6 | 71.5 | 79.7 | 63.8 | 60.9 | 66.7 |
| Ferritin (ng/mL) | 254.8 | 218.3 | 291.3 | 91.5 | 81.4 | 101.6 |
| Glucose (mmol/L) | 4.69 | 3.59 | 5.79 | 4.75 | 3.55 | 5.95 |
| TRSF (g/L) | 2.45 | 2.05 | 2.85 | 2.85 | 2.55 | 3.15 |
| Zn (µmol/L) | 39.9 | 38.3 | 41.5 | 14.8 | 14.0 | 15.6 |
| Cu (µmol/L) | 37.8 | 25.9 | 39.7 | 19.1 | 18.2 | 20.0 |
| Se (nmol/L) | 114.8 | 101.3 | 128.3 | 629.6 | 597.5 | 661.7 |
| SOD (µg/mL) | 8.1 | 6.9 | 9.3 | 20.4 | 17.9 | 22.9 |
| GPX (pg/mL) | 10.5 | 9.6 | 11.4 | 37.4 | 35.3 | 39.5 |
Serum iron and TIBC concentrations were increased in AD patients (41.4 µmol/L, and 75.6 µmol/L), compared to healthy controls (20.6 µmol/L, and 63.8 µmol/L); P<0.005. Plasma Se concentrations were decreased in AD (114.8 nmol/L) collate to control group (629.6 nmol/L); P<0.001. As an evidence of oxidative stress, SOD and GPX in alzheimer’s disease cases were decreased (8.1 µg/L, and 10.5 pg/mL) in comparison to healthy controls (20.4 µg/L, and 37.4 pg/mL); P<0.005. We establish increased serum Zn and Cu levels in AD patients (39.9 µmol/L, and 37.8 µmol/L); in healthy controls they were 14.8 and 19.1 µmol/L; P<0.001). Ferritin as a sign of elevated iron showed elevated concentrations in neurodegenerative disease (254.8 ng/mL), compared to healthy controls (91.5 ng/mL); P<0.005.
We found statistically different results in included groups for quantified serum hepcidin levels. The results obtained are presented in figure 1.
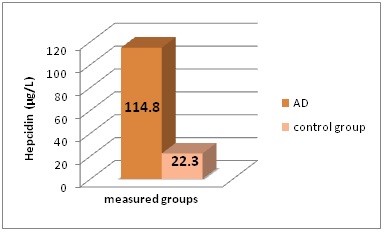 Figure 1: Serum hepcidin concentrations (in μg/L) in the different groups of patients included in this study.
Figure 1: Serum hepcidin concentrations (in μg/L) in the different groups of patients included in this study.
*AD - Alzheimer’s Disease
SOD and GPX levels statistically significant differs in AD patients and control group (P<0.005) (Figure 2 and Figure 3).
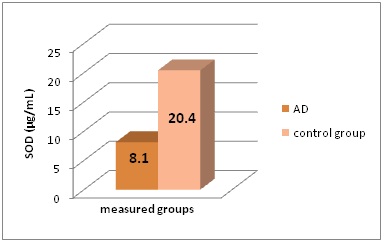 Figure 2: SOD levels (in μg/mL) in the different groups of patients included in this study.
Figure 2: SOD levels (in μg/mL) in the different groups of patients included in this study.
*AD - Alzheimer’s Disease, SOD - Superoxide Dismutase
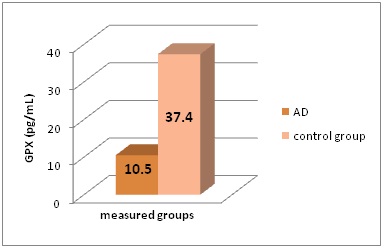 Figure 3: GPX levels (in pg/mL) in the different groups of patients included in this study.
Figure 3: GPX levels (in pg/mL) in the different groups of patients included in this study.
*AD - Alzheimer’s Disease, GPX - Glutathione Peroxidase
Serum hepcidin levels statistically differ between Alzheimer’s disease patients and healthy controls - 114.8 ± 10.1 µg/L to 22.3 ± 1.2 µg/L; P<0.001.
We found a significant correlation between serum iron levels and hepcidin concentration in included groups: patients with AD (r=0.698, P<0.001), and in control group (r=0.423, P<0.001). Ferritin levels correlated to quantified serum hepcidin in AD patients (r=0.544, P<0.005), and in control group (r=0.491, P<0.01). Plasma levels of Se correlated to hepcidin in AD cases (r=-0.781, P<0.005). Serum Zn and Cu levels correlated to hepcidin concentration in neurodegenerative disease (r=0.771 and r=0.698, P<0.005). SOD and GPX correlated to hepcidin in patients with Alzheimer’s disease (r=-0.825 and r=-0.865, P<0.005).
DISCUSSION
Balance between production and elimination of ROS is essential for maintaining its physiological level and the normal functions of the body. ROS are generated at different locations in the cell, especially in mitochondria. There enzymatic and non-enzymatic systems maintain constant levels of ROS. At low levels of ROS, the body cannot perform certain physiological functions. Conversely, high levels of ROS, leading to functioning in conditions of oxidative stress.
In oxidative stress, high ROS levels change all bio-macromolecules, causing lipid peroxidation, impaired transcription of RNA and DNA damage with the occurrence of mutations. The role of ROS in the oxidation of proteins is the basis for neurodegenerative diseases. ROS mediate and/or cause neurotoxicity. Free radicals pass through the plasma membrane and lead to cell death. ROS leads to the aging of the organism.
The most striking effect of aging can be described in neurodegenerative diseases associated with disturbed metabolism of microelements. Upon aging in the brain was observed accumulation of redox-metals (copper, iron and zinc). This occurs because of increased concentration of metals over passing Blood-Brain Barrier (BBB) in connectivity of neuronal medium with blood vessels. This makes the brain more likely to develop neurodegeneration [9].
AD is characterized by deposition of amyloid plaques of Amyloid-β chelating peptide (Aβ) with transition metal ions (Cu2+, Zn2+ and Fe3+). The binding of Cu2+ and Fe3+ leads to toxic chemical reactions, a change in the oxidation of two metals, the production of H2O2 in the presence of transition metals and finally gives toxic free OH radicals [2].
Some studies demonstrates role of oxidatively modified lipids in AD pathogenesis [10]. Lipid peroxidation mediated by ROS, generates reactive carbonyl ingredients, which can react with cellular proteins, lipids, and DNA leading to DNA damage. In vitro conditions, authors represents that the copper and iron ions presence catalyzes the formation of hydrogenperoxide [12]. More in vitro models shows that chelation of copper and iron inhibits ROS formation. The role of zinc in AD etiology is impressive - zinc inhibits Aβ-induced toxicity [11,12]. Additionally, zinc is connected to copper in alzheimer’s disease. While low zinc levels protects against Aβ-toxicity, its excess might lead to neuronaldeath. Under normal physiological conditions a sensitive balance exists between zinc, copper and Aβ-metabolism [13].
Neurodegenerative diseases associated with oxidative stress are not only caused by abnormal metabolism of trace elements, but there are genetic evidence suggesting that those associated with certain genetic mutations are more likely to develop nerve pathology compared to those with normal genetic profile. Patients with mutations in the HFE gene (for hemochromatosis) are more likely to develop oxidative stress and iron overload, leading to neuropathology [14,15].
Hepcidin is a key regulatory protein that controls the intestinal iron absorption and its distribution in the body [16]. It is a peptide hormone which is synthesized in the liver in response to such signals, including the levels of iron in the body. Hepcidin functions by binding and initialization degradation molecule ferroportin, the only known iron exporter. Ferroportin is located in the transmembrane duodenal enterocytes, macrophages and hepatocytes. It regulates the transfer of iron in plasma cell by these cells [17,18]. High levels of iron in the circulation stimulate the synthesis of hepcidin in the liver. Increased hepcidin reduces intestinal iron absorption and blocking exports by tissue depots-thus avoiding over-accumulation of iron in the body. On the other hand, decreased iron levels inhibit the synthesis of hepcidin and stimulate the absorption of iron in duodenal enterocytes and his release from tissue depots. Some researchers found decreased brain hepcidin deposition [19]. We quantified iron homeostasis regulator in serum.
Elevated hepcidin leads to decreased transferrin bound iron [20]. This is suggested to cause age-related anaemia [21].
CONCLUSION
Our study has some limits. The most important is the number of included patients. Still, we think our study contributs in practical introduction of quantification of serum hepcidin as a potential marker for early diagnosis of impaired iron homeostasis, leading trace element in the pathogenesis of neurodegenerative diseases. Increased serum hepcidin levels in combination with early manifestations of impaired information processing might be a useful guideline for focusing on early Alzheimer’s disease manifestations.
ACKNOWLEDGMENT
This project is implemented with the financial support of the Medical University - Sofia, as part of Grants - Project 5070/2016 (Contract 4-C/2016), and Contract 1-C/2015 (Project 4608/2015). We also kindly appreciate the support from “Marvena” (a “Siemens Healthcare Diagnostics” distributor in Bulgaria), especially to Mrs. Silviya Kirova and Mr. Velizar Dragoev.
REFERENCES
- Takahashi S, Takahashi I, Sato H, Kubota Y, Yoshida S, et al. (2001) Age-related changes in the concentrations of major and trace elements in the brain of rats and mice. Biol Trace Elem Res 80: 145-158.
- Opazo C, Huang X, Cherny RA, Moir RD, Roher AE, et al. (2002) Metalloenzyme-like activity of Alzheimer’s disease β-amyloidamyloid. Cu-dependent catalytic conversion of dopamine, cholesterol, and biological reducing agents to neurotoxic H(2)O(2). J Biol Chem 277: 40302-40308.
- Lakhal S, Schoedel J, Townsend AR, Pugh CW, Ratcliffe PJ, et al. (2011) Regulation of type II transmembrane serine proteinase TMPRSS6 by hypoxia-inducible factors: new link between hypoxia signalling and iron homeostasis. J Biol Chem 286: 4090-4097.
- Mastrogiannaki M, Matak P, Keith B, Simon MC, Vaulont S, et al. (2009) HIF-2alpha, but not HIF-1alpha, promotes iron absorption in mice. J Clin Invest 119: 1159-1166.
- Peeters-Scholte C, Koster J, Veldhuis W, van den Tweel E, Zhu C, et al. (2002) Neuroprotection by selective nitric oxide synthase Inhibition at 24 hours After Perinatal Hypoxia-Ischemia. Stroke 33: 2304-2310.
- Aki HS, Fujita M, Yamashita S, Fujimoto K, Kumagai K, et al. (2009) Elevation of jugular venous superoxide anion radical is associated with early inflammation, oxidative stress, and endothelial injury in forebrain ischemia-reperfusion rats. Brain Res 1292: 180-190.
- Viatte L, Lesbordes-Brion JC, Lou DQ, Bennoun M, Nicolas G, et al. (2005) Deregulation of proteins involved in iron metabolism in hepcidin-deficient mice. Blood 105: 4861-4864.
- Nicolas G, Chauvet C, Viatte L, Danan JL, Bigard X, et al. (2002) The gene encoding the iron regulatory peptide hepcidin is regulated by anemia, hypoxia, and inflammation. J Clin Invest 110: 1037-1044.
- Ceriello A (2000) Oxidative stress and glycemic regulation. Metabolism 49: 27-29.
- Hung YH, Bush AI, Cherny RA (2010) Copper in the brain and Alzheimer’s disease. J Biol Inorg Chem 15: 61-76.
- Cuajungco MP, Goldstein LE, Nunomura A, Smith MA, Lim JT, et al. (2000) Evidence that the beta-amyloid plaques of Alzheimer’s disease represent the redox-silencing and entombment of abeta by zinc. J Biol Chem 275: 19439-19442.
- Garai K, Sahoo B, Kaushalya SK Desai R, Maiti S (2007) Zinc lowers amyloid-beta toxicity by selectively precipitating aggregation intermediates. Biochemistry 46: 10655-10663.
- Cuajungco MP, Fagét KY (2003) Zinc takes the center stage: its paradoxical role in Alzheimer’s disease. Brain Res Brain Res Rev 41: 44-56.
- Gordeuk V, Mukiibi J, Hasstedt SJ, Samowitz W, Edwards CQ, et al. (1992) Iron overload in Africa. Interaction between a gene and dietary iron content. N Engl J Med 326: 95-100.
- Hely MA, Fung VS, Morris JG (2000) Treatment of Parkinson’s disease. J Clin Neurosci 7: 484-494.
- Ganz T (2011) Hepcidin and iron regulation, 10 years later. Blood 117: 4425-4433.
- Ramey G, Deschemin JC, Durel B, Canonne-Hergaux F, Nicolas G, et al. (2010) Hepcidin targets ferroportin for degradation in hepatocytes. Haematologica 95: 501-504.
- Sun CC, Vaja V, Babitt JL, Lin HY (2012) Targeting the hepcidin-ferroportin axis to develop new treatment strategies for anemia of chronic disease and anemia of inflammation. Am J Hematol 87: 392-400.
- Myhre O, Utkilen H, Duale N, Brunborg G, Hofer T (2013) Metal Dyshomeostasis and Inflammation in Alzheimer’s and Parkinson’s Diseases: Possible Impact of Environmental Exposures. Oxidative Medicine and Cellular Longevity 2013.
- Drakesmith H, Prentice AM (2012) Hepcidin and the iron-infection axis. Science 338: 768-772.
- Besson-Fournier C, Latour C, Kautz L, Bertrand J, Ganz T, et al. (2012) Induction of activin B by inflammatory stimuli up-regulates expression of the iron-regulatory peptide hepcidin through Smad1/5/8 signaling. Blood 120: 431-439.
Citation: Victor M, Savina H, Julia P, Vasil V, Maria P, et al. (2017) The Role of Iron Homeostasis in Alzheimer’s Disease. J Alzheimers Neurodegener Dis 3: 011.
Copyright: © 2017 Manolov Victor, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.