The Roles of Bacterial Products, Lipopolysaccharides and Bacteriocins, in Obesity and its Related Complications
*Corresponding Author(s):
Hamid Reza KazeraniAssociate Professor, Department Of Physiology, The School Of Veterinary Medicine, Ferdowsi University Of Mashhad, Mashhad, Iran, Islamic Republic Of
Tel:+98 51 38805632,
Email:kazerani@um.ac.ir / kazrani@yahoo.co.uk
Abstract
The expanding problem of obesity is becoming a very serious issue in developed and even developing societies. Humans have never been exposed to this level of food availability before the beginning of the modern era. Lifestyle change from active to sedentary form has increased in technology ruling period. Even the children are not in the safe margin, with ever increasing cases of overweight and obese children seen on the streets. In this review, we will consider obesity with special attention to adipocyte types, microbiota, and microbial products, LPS and bacteriocins.
Keywords
Adiposity; Bacteriocins; Lipopolysaccharide; Microbiota; Obesity; Overweight
Introduction
Obesity is a growing problem of modern societies. According to the World Health Organization (WHO) report in February 16th2018, obesity has nearly tripled since 1975.According to WHO reports, in 2016, more than 1.9 billion adults, 18 years and older, were overweight, more than 650 million of which were affected by obesity. Obesity is also increasing among children. At the moment of writing this article, 41 million children of age 5 or less are affected by obesity in the world [1]. A survey of people with obesity in the US shows that 35% of men and 40.4% of women are suffering from obesity [2]. The scale used to determine obesity is mostly Body Mass Index (BMI). The BMI is calculated as body weight in kilograms divided by the square of height in meters (kg/m2). World Health Organization defines the BMI of 25 or higher as overweight, whereas that of 30 or higher would be obese.
Complications related to obesity
Obesity-related complications including Diabetes Mellitus type II (T2DM), cardiovascular diseases, hypertension, metabolic syndrome, asthma, gastrointestinal problems, skeletal disorders, and even cancer are becoming epidemic [3]. Depression and obesity are strongly interrelated: The occurrence of one increases the risk of the other [4].
Obesity and non-insulin dependent diabetes mellitus: Obesity is a prominent risk factor for Non-insulin Dependent Diabetes Mellitus, (NIDDM), which is also called diabetes mellitus type II. In a study of 8430 men and 7034 women affected by obesity, it was shown that people with ectopic fat obesity are in the greatest risk of NIDDM [5]. Ectopic obesity is defined as the accumulation of triglycerides in non-adipose tissues [6]. Insulin decreases the proliferation of T-cells; in addition, obesity increases the production of IL-1β, which may have cytotoxic effects on beta islet cells [7]. Insulin resistance is present in NIDDM patients 10 to 20 years prior to disease onset. Increased plasma free fatty acid concentration is associated with insulin resistance. Moreover, there is a direct relationship between intra myocellular triglyceride accumulation and insulin resistance. Some studies suggest that an increase in plasma fatty acid concentration reduces glucose uptake and phosphorylation, and this leads to reduced glycogen synthesis and glucose oxidation. In addition, there is evidence that fatty acids interfere with very early stages of Glucose Transporter Type 4 (GLUT4) and hexokinase II activity [8]. Since carnitine is involved in fat metabolism, attention is being paid to its possible role in NIDDM. Carnitine plays a role in the transfer of long chain fatty acids from cytoplasm to mitochondria for beta-oxidation. L-carnitine, in addition, has anti-inflammatory and antioxidant properties [9]. Carnitine can improve insulin sensitivity too [10]. One theory suggests that lipid over supply leads to defect in Insulin Receptor Substrate (IRS) through serine/threonine phosphorylation. It is believed that theproduction of ceramide can reduce insulin efficiency via long chain acyl-CoA. In addition, mitochondrial stress is caused by accumulation of incompletely metabolized lipids, which leads to insulin resistance. L-carnitine supplementation decreases serum glucose but has no effect on glucose oxidation [11].
Obesity and cardiovascular complications: Cardiovascular diseases are another cause of ill health in modern societies. Obesity is associated with an earlier onset and increased risk of cardiovascular disorders including hypertension [12]. Atherosclerosis caused by increased serum triglyceride and cholesterol is a major cause of hypertension in obese individuals. In addition, obese individuals have a higher concentration of norepinephrine in their serum. It seems that this increase is due to high-fat diet, and ultimately stimulation of alpha-1 and beta-adrenergic receptors [13]. Hyperglycemia related to obesity might have adverse effects on endothelium, vascular smooth muscle cells, and macrophages, leading to thrombosis, fibrinolysis, and finally atherosclerotic plaques. Moreover, hyperglycemia causes overproduction of Reactive Oxygen Species (ROS), which is a cause of low-grade inflammation and Cardiovascular Diseases (CVD). Patients with NIDDM die three to four times more from CVD than normal individuals. Hypertrophy of visceral adipose tissue causes cell hypoxia and apoptosis, which attracts macrophages and causes a low-grade inflammation [14].
Obesity and metabolic syndrome: Metabolic syndrome, a complication of obesity, is closely associated with increased cancer risk, especially prostate cancer [15]. Metabolic syndrome is a collection of complications including central obesity, increased serum glucose and lipids, and high blood pressure. Other than cancer, in this syndrome, there is an increased risk of cardiovascular diseases and NIDDM [16]. In addition, metabolic syndrome is connected with Helicobacter pylori infection and its associated complications. It seems that H. pylori infection and gastric and duodenal ulcers increase the occurrence of metabolic syndrome [17], but the opposite is not established. In addition, metabolic syndrome is positively correlated with some urological disorders such as hypogonadism, nephrolithiasis, overactive bladder, and erectile dysfunction. Fasting plasma insulin has been connected to Benign Prostatic Hyperplasia or BPH. Based on this, increased plasma insulin is a factor that can promote both BPH and prostate cancer [18]. Some of the very serious diseases of the liver such as non-alcoholic fatty liver disease, Hepatocellular Carcinoma (HCC), and generally liver-related mortality are the complications of metabolic syndrome [19].
Obesity and asthma: Asthma, which is characterized by recurrent attacks of breath shortness, cough, and wheeze, is caused by airway inflammation. More than 300 million people nowadays are affected by asthma. Asthma and obesity are increasing in a parallel way. Actually, the prevalence of obesity is more among asthma affected individuals. People with asthma, which are at the same time obese, show more resistance to the classical treatment of asthma. The prevalence of obese asthma is more among females. It seems that inflammation associated with obesity, which is caused by cytokines of adipose tissue, may participate in the onset of asthma [20]. Asthmatic patients have higher serum levels of leptin and lower levels of adiponectin. Leptin is a pro-inflammatory adipokine and is produced by adipose tissue, and its serum levels are higher in obese individuals. On the other hand, adiponectin is an anti-inflammatory agent and it reduces pro-inflammatory cytokines like Tumor Necrosis Factor alpha (TNF-α) and Interleukin-6 (IL-6) [21].
Obesity and cancer: In addition to well-known causes of cancer such as smoking, pollution, stress, etc., now there is considerable attention toward obesity. It seems that the low-level constant inflammation in obese cases is a key factor in the increased occurrence of cancer. Kynurenine pathway is the primary pathway of tryptophan catabolism in the liver, and it is the point where the synthesis of Nicotine Amideadenine Dinucleotide (NAD+) begins. This pathway is important in inflammation [22]. Different studies suggest that between 5.5 to 14% of cancer cases are related to obesity. An increase in unbound Insulin-like Growth Factor-1 (IGF-1) and insulin in obese people who have NIDDM symptoms may lead to tumor development. High levels of insulin reduce hepatic production of IGF Binding Protein (IGFBP), which leads to increased free serum IGF-1. Increased IGF-1 and its receptor are closely associated with colorectal cancer. Both insulin and IGF-1 receptors promote phosphoinositide 3-kinase (PI3 kinase)/protein kinase B cascade, and this prevents apoptosis pathways. Another obesity-related factor that contributes to cancer is leptin, a hormone of adipose tissue, which increases the risk of colorectal and breast cancers. Leptin is a cell proliferation promotor and its receptors are present on several types of cancer cells. It promotes cyclin D1 and suppresses apoptosis. Leptin increases PI3K and Mitogen-Activated Protein Kinase (MAPK) which leads to angiogenesis and tumor growth. Normally, leptin is considered a satiety hormone by acting on hypothalamus arcuate nucleus [23].
As adipocytes increase in size due to increased lipid production, some of them become apoptotic and surrounded by immune cells. Both adipocytes and immune cells produce inflammatory cytokines which can cause a low-grade inflammation. Mitochondrial dysfunction and endoplasmic reticulum stress alter cytokine production. Interleukin-4 (IL-4) from adipose tissue is an anti-inflammatory cytokine which reduces inflammation through type 2 helper (TH2) cells and regulatory T cells (T-Regs). Chronic obesity causes a reduction in TH2 and T-Reg population and an increase in pro-inflammatory TH1 and CD8+ T-cells. The cytokines IL-6, IL-11, TNFα, IL-1β, IL-23 are tumor promoting, and there is evidence that at least some of them can increase in obese cases. In fact, adipose tissue, other than being an energy storage organ, is an important endocrine organ regulating metabolism and inflammation. Adiponectin, a product of adipose tissue, has been shown to exert anti-cancer effects by attenuating inflammation and cell proliferation; however, it is reduced in obesity and so is increased the risk of cancer and systemic inflammation. Osteopontin, a phosphoprotein expressed by adipose tissue, can increase the survival and metastasis of neoplastic cell [24]. Numerous other factors areinvolved in this process too,which are not the main concern of this text, but just to give hints.
The Causes of Obesity
Several factors contribute to the prevalence of obesity which can be divided into genetic and environmental factors.
Several genes have been shown to be involved in monofactorial obesity [25]. These include the leptin gene, Melanocortin 4 Receptor (MC4R), Brain Derived Neurotrophic Factor (BDNF), Proprotein Convertase Subtilisin/kexin type 1 (PCSK1), Adrenoceptor Beta 3 (ADRB3), and peroxisome proliferator activated receptor gamma (PPARγ) genes. Sex has a direct effect on the distribution and the amount of body fat [26]. Twenty loci on chromosomes have been attributed to obesity [27]. These loci control the amount of food intake in the CNS and fat metabolism. Many of these genes in the CNS work in conjunction with leptin, so the appetite/satiety pathway is disturbed, resulting in overeating. Some of these effects are syndromic, which are not of our interest. However, many of them are related to common obesity. Many of these genes are highly expressed in the hypothalamus, where appetite is set. An increase in consumption of high caloric foods, a decrease in physical activities, an increase in sedentary jobs, and as a whole the decreased calorie consumption can be classified as environmental factors [28].
Adipose Tissue And Fat Cell Types
Mammalian fat tissues are classified as White Adipose Tissue (WAT), Brown Adipose Tissue (BAT) and beige (brite) fat, with the latter being interspersed among WAT cells. White adipose tissue has the main job of energy storage. Based on the distribution pattern of fat tissue, there are two main patterns of obesity: android and gynoid. According to the definition, android obesity is characterized by fat accumulation in the abdominal region, however, in gynoid pattern accumulation is in the hip and thigh area, mostly subcutaneous. According to the literature, android obesity has a higher correlation with cardiovascular and metabolic disorders, while gynoid obesity is more protective [29]. White adipose tissue cells contain large droplets of fat in the cytoplasm. This tissue can be considered an endocrine organ, secreting hormones such as leptin, adipsin, Acylation-Stimulating Protein (ASP), angiotensinogen, Plasminogen Activator Inhibitor-1 (PAI-1), adiponectin, resistin, some steroid hormones and several cytokines [30].
White adipose tissue grows by either hypertrophy or hyperplasia. What determines either way, is genetic and environmental factors [31]. In humans who have adipose tissue hypertrophy, there is more frequency of NIDDM, suggesting that hyperplasia might be protective against at least diabetes [32]. The distribution of adipose tissue is different between male and female. Men tend to accumulate fat more in abdominal areas, typical of android obesity, which is more associated with cardiovascular disorders, while women tend to have more subcutaneous (gynoid) obesity, which is more protective against cardiac diseases [33]. This difference is attributed to estrogens actions.
It was previously thought that Brown Adipose Tissue (BAT) disappeared after infancy, but now we know that it exists in adult life and is gaining more importance in the etiology of obesity. Its level of activation during adulthood has a dramatic inverse correlation with the incidence of obesity. Brown adipose tissue contains less amount of fat compared to WAT, and it “burns” this depot to produce heat; it is highly vascularized and receives sympathetic innervation, being stimulated by beta-adrenergic stimulation. It has more mitochondria compared to WAT. Uncoupling Protein-1 (UCP-1) is highly expressed in the mitochondrial membrane of BAT. Instead of conducting protons toward ATP synthase to produce ATP, it allows protons to leak through and produce heat (Figure 1). This is increased by beta-adrenergic stimulation [34]. In humans, BAT is mostly found in the cervical region. Brown adipose tissue cells store fat in a multilocular form. Brite or beige adipocytes, which are called inducible brown adipocytes too, are interspersed among WAT cells and look like WAT. Normally, the proteins and patterns of gene expression in brite cells resemble those of WAT. However, when stimulated by the cold or beta-adrenergic system, they become multinuclear and express UCP-1 [35]. It is now believed that WAT can be converted to brite tissue, and this may have a significant effect on body weight.
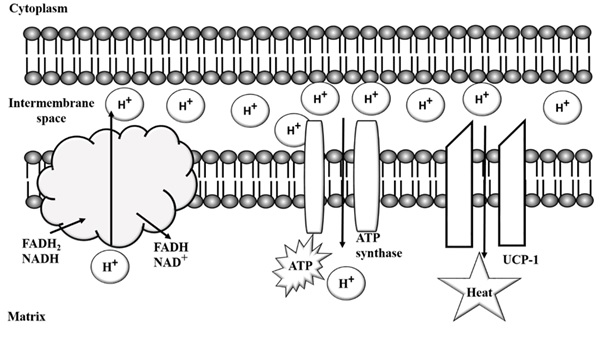 Figure 1: Brown adipocyte mitochondrion containing Uncoupling Protein-1 (UCP-1). FADH2: Flavin Adenine Dinucleotide; NADH: Nicotinamide Sdenine Dinucleotide.
Figure 1: Brown adipocyte mitochondrion containing Uncoupling Protein-1 (UCP-1). FADH2: Flavin Adenine Dinucleotide; NADH: Nicotinamide Sdenine Dinucleotide.
Origins of WAT, BAT and brite cells
White adipose tissue cells first appear in stromal vascular formation with a CD45-CD31- Ter119- (Lin-) CD29+Sca1+CD34+CD24+ pattern (Figure 2). Peroxisome Proliferator-Activated Receptor Gamma (PPARγ) is an early WAT commitment marker, and adipose vascular mural cells are the source and niche of WAT. Actually, there is a hypothesis that WAT cells arise from a subset of endothelial cells [35]. The transcriptional factor Zinc Finger Protein 423 (zfp423) is a key regulator of WAT maintenance, suppressing the thermogenic mechanisms in brown and brite adipose tissues. If zfp423 is suppressed by a combination of doxycycline and beta-adrenergic stimulation, WAT cells will start showing the characteristics of brite cells. Early B-cell Factor 2 (Ebf2) and PR Domain containing 16 (prdm16) are two transcriptional regulators that establish brown and brite adipocyte traits, and zfp423 acts as a suppressor to these two, guaranteeing the maintenance of WAT trait [36]. Brown adipocytes are derived fromthe Myf5+ lineage of mesenchymal stem cells, where brite and white adipocytes are from Myf5- lineages (Myf5: myogenic factor 5) [37].
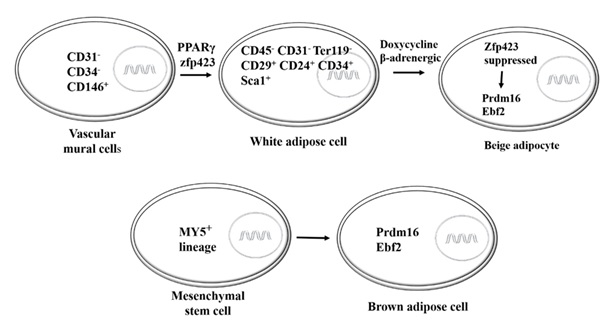 Figure 2: The origin of adipocytes. CD: Cluster of Differentiation; PPARγ: Peroxisome Proliferator-Activated receptor gamma; zfp423: Zinc Finger Protein 423; Prdm16: PR Domain Containing 16; Ebf2: Early B cell Factor2.
Figure 2: The origin of adipocytes. CD: Cluster of Differentiation; PPARγ: Peroxisome Proliferator-Activated receptor gamma; zfp423: Zinc Finger Protein 423; Prdm16: PR Domain Containing 16; Ebf2: Early B cell Factor2.
Peroxisome proliferator-activated receptor γ coactivator 1α (PGC1α) is the main factor in regulating UCP1 related thermogenesis in BAT. This factor induces UCP1 expression. In addition, PGC1α regulates Nuclear Respiratory Factors 1&2 (NRF1 and NRF2), which themselves are regulatory transcription factors modulating the expression of genes encoding respiratory chain subunits [38].
Adipose tissue secretions: Adipokines
Previously, it was thought that adipose tissue was just an organ for saving excess energy, in other words, it was a deposit. However, now we know that it is far more than just that. Adipose tissue is a large endocrine organ with various secretory chemicals, affecting different aspects of homeostasis ranging from food intake and insulin resistance to cardiovascular diseases, immunity, and inflammation, coagulation and reproduction. Many of these secretions are called adipokines. Secretory nature of adipose tissue was noticed in 1994 by the discovery of leptin. Now we know that numerous products are made in adipose tissue, to mention a few would be IL-6, TNF-α, sex steroids, adiponectin, glucocorticoids, visfatin, resistin, adipsin, and angiotensin. In (Table 1), a brief description of known adipose tissue secretions is listed [39].
|
Factor |
Description |
|
Leptin |
Described in 1994, pro-inflammatory, anorexigenic, increases energy usage, cleared from plasma by filtration, degraded in renal tubules, receptors in CNS and many other tissues, higher in obese people, higher in females |
|
Adiponectin |
Described in 1995, has three isoforms of trimeric, hexameric and multimeric forms, its plasma concentration has a negative correlation with fat mass, improves insulin sensitivity, stimulates fatty acid oxidation and glucose uptake in skeletal muscle, suppresses hepatic glucose output |
|
TNF-α |
From both adipose tissue and macrophages that infiltrate the tissue, it can impair insulin signaling in hepatic and adipose tissue, it reduces fatty acid oxidation in hepatocytes |
|
IL-6 |
30% of circulating IL-6 originates from adipose tissue, higher in visceral fat, increase with obesity and plasma free fatty acids, it inhibits insulin signaling pathway |
|
Angiotensin |
Decreases in fasting, stimulates prostacyclin synthesis, adipocyte differentiation, and lipogenesis |
|
PAI-1 |
More in visceral than subcutaneous adipose tissue, seems to be involved in cardiovascular risks |
|
ASP |
Increases lipogenesis, it increases with meals and facilitates the storage and synthesis of triglycerides, increases by obesity and decreases by weight loss, it is increased by cardiovascular diseases and decreased by age |
|
Resistin |
Secreted by a large number of cells, increases by obesity, has a negative effect on insulin sensitivity, is involved in diabetes complications, it leads to insulin resistance in fat tissue, it increases hepatic gluconeogenesis |
|
Visfatin |
Prevalent in visceral fat tissue, is involved in B-cell proliferation and differentiation of pre-adipocyte to adipocyte, pro-inflammatory and is involved in innate immunity, regulates cellular levels of NAD+ and so cell metabolism, it binds to and activates insulin receptor decreasing plasma insulin |
Table 1: Some factors secreted by adipocytes TNF-α: Tumor Necrosis Factor alpha; IL-6: interleukin 6; PAI-1: Plasminogen Activator Inhibitor-1; ASP: Acylation Stimulating Protein. Please refer to [39], for more information.
Microbiota
The human gut is host to more than 3.8 ×1013bacterial cells, not to mention viruses and prokaryotes [40]. This population includes a wide variety of gram-positive and gram-negative species. These species have a wide range of activities from food digestion to immune modification. The population is lower in acidic upper Gastro Intestinal system (GI), but as we go down to the moderate niche of caecum and colon, the number and variety increase. Since the GI is one of the largest interacting surfaces of inside and outside the body, this population can be altered by different environmental and internal factors, and this alteration will have a clear effect on whole body status. This alteration seems more important when we consider that the amount of genetic content in these bacterial species in the gut is 100 times more than the human genome [41].
To refer to microbial content of the human gut, we use the two terms microbiota and microbiome. The whole population of micro-organisms in the GI is called microbiota, while its genetic content is called microbiome. Many of the bacterial species in the GI cannot be cultured by conventional microbiology media; just 30% of bacterial species can be cultured, so other techniques should be used to identify them. Many of these techniques work with 16S rRNA. Recently, the study of the whole genome from an environment, called metagenomics, has been applied to identify the population changes. Some other employed techniques include qPCR, DNA microarrays and microbiome shotgun sequencing [42].
The human infant gut does contain some bacterial species even before birth, but this population quickly increases after birth. Firmicutes are present in meconium, and as the time passes, proteobacteria appear in feces. However, the kids which are born with cesarean section have less similarity to their mothers in their gut microbiota compared to the kids born vaginally. Other factors contributing to the composition of human infant gut microbiota include breastfeeding, antibiotic therapy, host genes and environmental conditions [43]. As the person grows, there might be some changes in this population. The bacterial composition may vary from person to person, but generally, it mainly contains species of Bacteroidetes and Firmicutes.
One of the main factors affecting body weight is the microbiota of the GI [44]. More than 100 species inhabiting human GI are divided into two main groups: gram-positive and gram-negative bacteria. Alterations in these bacterial cells have important effects on body weight in rodents and humans. As an example, the administration of bacterial species Lactobacillus paracasei , L. rhamnosus, Bifidobacterium animalis subsp. lactis has shown a reduction in weight gain in mice fed with high-fat diet [45]. In another study, 14 Lactobacillus species were identified to be associated with weight changes in humans using bioinformatic analysis [46]. Based on this study, bacterial species L. reuteri, L. acidophilus, L. fermentum, L. sakei, L. ingluviei are classified as weight gain bacteria, while L. plantarum and L. gasseri are among weight loss bacteria.
The mechanisms underlying weight changes caused by the microbiota
Several mechanisms have been suggested regarding how microbiota can cause alterations in body weight. One is the production of Short Chain Fatty Acids (SCFAs) by bacteria. The undigested or unabsorbed carbohydrates in the form of starch or fiber are metabolized by microbiota into SCFAs. These products, mainly acetate, propionate and butyrate, can be used by the host intestinal cells [47]. Many bacterial species are involved in these pathways, such as Akkermansia muciniphilia, Faecali bacterium prausnitzii species of Bifidobacterium, Prevotella, Ruminococcus, Bacteroides, Salmonella, Eubacterium, and Roseburia as some examples. The SCFAs produced by these bacteria are also used to produce mucin by large intestinal goblet cells through activation of the protein MUC2 expression [48]. They can provide up to 10 percent of human energy requirements. Two G-protein receptors in the small intestine and colon cells, GPR41 and GPR43, are responsible for the metabolic effects of acetate, propionate, and butyrate. These receptors have been seen in other tissues such as adipose tissue, mononuclear white blood cells, lymph nodes, spleen, heart and skeletal muscle cells [49].
Appetite in humans is controlled by the CNS, through hypothalamus and brainstem. The GI microbiota can regulate the signalling patterns to the CNS through vagus pathways, modulating the GI motility and satiety [50]. Bacterial species in the human gut can produce several neuroactive factors, which are able to affect gut-brain axis, some of which are Gamma-Aminobutyric acid (GABA) from Lactobacillus and Bifidobacterium strains, histamine from Lactobacillus reuteri, and acetylcholine produced by both bacteria and fungi. There is evidence suggesting that gut microbiota can alter dopamine, epinephrine, norepinephrine and serotonin levels [51].
Lipopolysaccharide: Lipopolysaccharide (LPS) is a structural element of the gram-negative bacterial outer membrane. It is composed of three structural parts: lipid A, polysaccharide chain and antigen O (Figure 3). The diversity of antigen O determines different serological activities of LPS types.
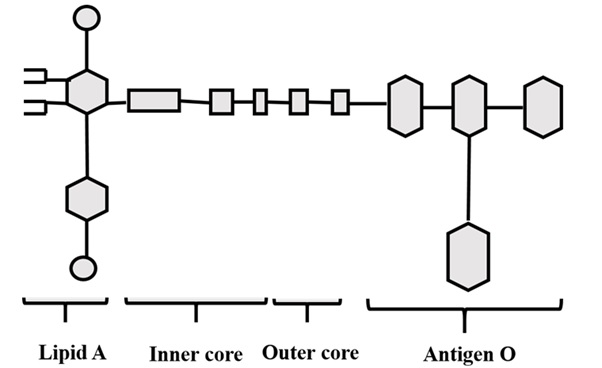 Figure 3: The chemical structure of LPS.
Figure 3: The chemical structure of LPS.
Lipopolysaccharide is detected by Toll-like Receptor 4 (TLR4) which attaches to Lipopolysaccharide-Binding Protein (LBP) bound LPS and subsequently to CD14. In other words, LPS binds to serum protein LBP, and LBP then catalyzes the transfer of the LPS to serum or membrane-bound CD14. Finally, CD14-LPS complex interacts with TLR4, leading to signal transduction across the membrane [52]. TLR4 is expressed on a wide variety of cells, but the immune response to LPS is basically on innate immune cells (Figure 4).
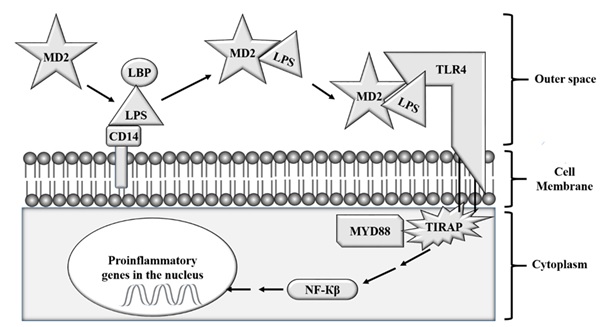 Figure 4: The mechanism of LPS action. TLR4: Toll-Like Receptor 4; MD2: Lymphocyte antigen 96; LBP: LPS binding protein; CD14: Cluster of Differentiation 14; MYD88: Myeloid Differentiation Primary Response 88; NF-κβ: nuclear factor kappa beta; TIRAP: Toll-Interleukin 1 receptor domain containing adaptor protein.
Figure 4: The mechanism of LPS action. TLR4: Toll-Like Receptor 4; MD2: Lymphocyte antigen 96; LBP: LPS binding protein; CD14: Cluster of Differentiation 14; MYD88: Myeloid Differentiation Primary Response 88; NF-κβ: nuclear factor kappa beta; TIRAP: Toll-Interleukin 1 receptor domain containing adaptor protein.
Lipopolysaccharide molecules are important in medicine because they are the main cause of septic shock caused by severe gram-negative bacterial infection. However, a chronic state of elevated LPS levels in the blood can cause hyperglycemia, decreased body fat mass, increased liver weight and increased pro-inflammatory cytokines in frontal cortex [53,54], showed that administration of LPS at 100ng/ml to 3T3-L1 preadipocytes reduces adipogenesis in the cells. Neutral fats can be measured by oil red O staining in the cells and this method is used to check the appearance of adipogenesis in adipose cells. It has been shown that the genes such as PPARγ and Adipocyte binding Protein 2 (aP2), which are master adipogenic regulators, and which are involved in fat production and adipocyte differentiation,are downregulated by LPS. In addition, LPS inhibits phosphorylation of AMP-Activated Protein Kinase (AMPK). A study on Stromal Vascular (SV) cells showed that LPS administration induces early senescence in the cells; moreover, it activates P53 and increases TNF-α, IL-1, IL-6, Monocyte Chemoattractant Protein-1 (MCP-1) and Vascular Endothelial Growth Factor (VEGF) secretion [55]. These effects are independent of NF-kB.
Contrary to the above-mentioned studies, some researchers suggest that LPS can induce obesity, insulin resistance and diabetes [56]. In these conditions, sub clinical LPS levels is an inducer of low-grade chronic systemic inflammation. According to [56], high-fat diet can increase serum LPS levels up to 4 times due to the rise in the ratio of LPS containing bacteria in the gut. On the other hand, when LPS is injected subcutaneously, the whole body fat is increased just similar to that observed in the mice fed with high-fat diet. The reason behind this controversy between the resultson cell-line studies compared to in vivo trials is not clear. The prolonged exposure to low levels of LPS (metabolic endotoxemia) causes inflammation in hypothalamus through JNK pathway, making hypothalamus irresponsive to circulating insulin levels, a central resistance to insulin. This way, the hypophagic response to insulin fades, which can be a reason for obesity. Lipopolysaccharide itself can cause central hypophagia viaPhosphatidylinositol-4, 5-bisphosphate 3-kinase (PI3K) pathway, however, repeated exposure to LPS can cause a resistance to hypophagia and lead to obesity as mentioned above. On the other hand, acute endotoxemia promotes insulin secretion and activates insulin signaling cascades [57].
Bacteriocins: Bacteriocins are peptides produced by bacteria in their ribosomes. These peptides have different ranges of antibacterial activities [58]. They can be narrow-spectrum, effective against only limited types of bacteria, or broad spectrum, effective against a large number of bacterial species. For example, lactic acid bacteria, produce bacteriocins that are exclusively effective against gram-positive bacteria and especially other species of Lactobacillus [59]. Some bacteriocins like nisin are used in food industry as preservatives to protect against food spoilage. There have been efforts to determine the structure of some bacteriocins, but we don’t have sufficient data in this regard.
At the moment, there is no report that independently can verify the effect of bacteriocins on weight. Some studies have considered the proteins produced by bacteria and their effect on carbohydrate and lipid metabolism. According to one study, weight protecting bacteria produce more glucose permease and are more involved in defense against oxidative stress. Moreover, weight gain bacteria produce more bacteriocins than weight protection ones (Table 2). Weight gain associated bacteria lack enzymes related to fructose metabolism and the defense against oxidative stress [46].
|
Bacteriocins associated with weight gain |
Bacteriocins associated with weight loss |
||
|
L. acidophilus |
Gassericin |
L. plantarum |
Reutericin |
|
Enterolysin |
Enterosin |
||
|
Lactacin |
Plantaricin |
||
|
Carnocin |
UviB |
||
|
Nukacin |
|||
|
L. reuteri |
Paenicidin |
|
|
|
Colicin |
|||
|
L. sakei |
Aureocin |
|
|
|
Sakacin |
|||
Table 2: Lactobacillus bacteriocins assumed to be engaged in weight gain or weight loss.
Microbiota in Obesity-Associated Complications
Non-insulin Dependent Diabetes Mellitus (NIDDM)
Since NIDDM is associated with low-level chronic inflammation, resident GI microbiota which is related to chronic inflammation has been linked to the occurrence of NIDDM. Obese people who show the signs of insulin resistance have a higher ratio of Firmicutes/Bacteroidetes compared to normal people. There is evidence that changes like these make the intestine more permeable to bacterial toxins and may result in inflammation. It seems that some gut microbiota predisposesthe adipose tissue to low-level inflammation. This can lead to prediabetic situations like insulin resistance. Bacterial species such as Bacteroidetes thetaiotaomicron, Akkermansia muciniphila and Escherichia coli affect glycocalyx and mucus layers, altering intestinal permeability. Moreover, microbiota alters gut tight junctions, endocannabinoid system, and intestinal alkaline phosphatase activity, which can disturb intestinal permeability. The opportunistic pathogenic bacteria and some gram-negative endotoxin producing bacteria show overgrowth in NIDDM patients. Their LPS, flagellin, and peptidoglycan can be absorbed and cause the low-level inflammation. We know that Lactobacillus reuterican reduce serum levels of glucose and leptin, and adipose tissue IL-6 and TNF-α. Lactobacillus caseican increase serum LPS-Binding Protein (LBP) and reduce serum LPS levels [60]. Probiotics have an effect on reducing high-sensitivity C-reactive protein (hs-CRP) levels in serum. Among these probiotics we can refer to Lactobacillus casei, Bifidobacterium breve, Lactobacillus reuteri, Lactobacillus plantarum and Lactobacillus acidophilus [61].
Several glucose-lowering agents used in NIDDM patients have been found to alter gut microbiota both in positive and negative ways. There are theories that these agents might affect gene expression and short chain fatty acid production in bacteria and hence evoke cytokine production and inflammation. Metformin, a drug used to lower glucose in NIDDM patients which possesses pleiotropic effects, improves glucose uptake and usage by microbiota, increases Glucagon-Like Peptide-1 (GLP-1) and bile acid levels, and finally might alter the composition of microbiota. The finding that intravenous injection of metformin is not as effective in reducing serum glucose levels as its oral administration suggests a critical role for microbiota in the occurrence and treatment of NIDDM. Unfortunately, the microbial alteration caused by metformin might lead to intestinal intolerance. Another example can be GLP-1: Gut microbiota digest non-digestible carbohydrates and produce Short Chain Fatty Acids (SCFA) which bind to GPR41 and GPR43 receptors. This leads to GLP-1 production, which is a regulator of glucose metabolism, appetite and gastric emptying [62]. A carbose, which is recently being used in Asian countries in the place of metformin, is an inhibitor of alpha-glycosidase. As a result, the complex carbohydrates remain in the intestine and glucose absorption is reduced. A carbose treatment increases the ratios between primary and secondary bile acids and plasma levels of unconjugated bile acids through microbial alterations, and this might lead to metabolic improvements. A carbose increases the relative abundances of Lactobacillus and Bifidobacterium and reduces Bacteroides in the intestine, so it changes the relative abundance of microbial genes involved in bile acid metabolism [63].
Cardiovascular Diseases (CVD)
Blood pressure is regulated by several factors, including genetic, dietary, environmental and endocrine agents. Recently, there have been discussions around gut microbiota being able to cause hypertension. Hypertensive patients show signs of gut dysbiosis and SCFA alterations. Short chain fatty acids can regulate blood pressure by acting on renal GPR41, GPR43, and Olf78 receptors. Similar to obese individuals, the ratio of Firmicutes/Bacteroidetesis significantly higher in hypertensive animals. A family of Bacteroidetes, called Veillonellaceae seems to increase the sensitivity of rats to high salt diet and increase blood pressure. A derivative of gut microbiota, trimethylamine N-oxide (TMAO), is linked to atherosclerosis. Clostridiaceae and Peptostreptococcaceae families have been reported to be positively related to TMAO levels [64]. Atherosclerotic plaques contain the DNA from the same bacterial taxa present in patients’ guts [65].
Asthma
Asthma drugs show a reduced efficacy in obese people. There are discussions that obesity might alter liver microbiota. In obese asthmatic people, weight loss reduces asthma symptoms. The role of gut microbiota has not been verified in obesity-related asthma; evidence shows that this connection does exist. Innate immunity cells which have been presented by specific antigens in one mucosal site might migrate to other mucosal sites, exerting immune responses in the new area. Bacterial metabolites might alter immune responses in the lungs too [66]. There is evidence suggesting that SCFA might affect obesity-related asthma. Germ-free mice, which cannot produce SCFA as much as conventional mice, show a greater sensitivity of airways to allergic responses. Propionate (via administration or by high fiber diet) in mice has reduced allergic airway inflammation [67].
Cancer
Cancer is a leading cause of death in the world. Although there are different ideas about how cancer is related to obesity, here we intend to have a brief look at obesity-related microbiota and its connection with the risk of cancer. One possible underlying mechanism is the production of procarcinogenic factors by microbiota. As an example, the pathogenic species Bilophilawads worthia grows more when there is an abundance of taurocholic acid, which itself is a metabolite of milk saturated fat in the colon. This bacterial species is related to colitis, and colitis is a risk factor for colon cancer. As another example, Clostridium sordellii which grows in high-fat fed mice increases the production of deoxycholic acid, and this metabolite is a potential DNA damaging agent increasing the risk of hepatic cancer [68]. There is evidence suggesting a correlation between obesity and cancers of gallbladder, liver, thyroid, and ovary too. Excess body fat is associated with increased M1 macrophages, increased T-helper 1 cytokines (pro-inflammatory cytokines) and decreased T-helper 2 cytokines (anti-inflammatory cytokines). Elevated C-Reactive Protein (CRP) levels, which occurs in obesity, is associated with several cancer kinds, including colon and breast cancer. This inflammation is responsible for breast cancer recurrence too [69].
Bacterial species such as Bacteroides fragilis, Shigella spp., Citrobacter spp. and Salmonella sppcan accumulate in the intestinal epithelium, cause inflammation, produce cellular proliferation and lead to a pre-malignant state. When the tumor ruptures, the change in the environment causes a shift toward opportunistic species such as Fusobacterium species and some members of the Streptococcaceae family, which thrive due to barrier dysfunction. Fusobacterium nucleatum has been reported to be present in greater numbers in obese people, and there is evidence that it is involved in colorectal cancer [70].
The Future Horizon
Bacteria and bacterial products are showing significant roles in host metabolism. There are reports regarding the effect of LPS on the expansion of adipose tissue. However, the results were contradictory and open to study. How come direct LPS in cell culture can cause adiposity repression, while in vivo application can cause obesity and NIDDM? Is it just related to inflammation? Is it related to microbial niche alteration? Previous studies show that bacteriocins can cause alterations in bacterial carbohydrate metabolism. According to the preliminary studies, the interaction of bacteriocins with their receptors on bacterial cell wall can alter the cell metabolism [71]. Since bacteriocins have antibiotic effects, their presence in the GI can cause alterations in microbial populations, and since microbial populations are different in obese and lean people, bacteriocins might be able to cause weight changes. However, the possible direct effects which bacteriocins might have on adiposity and obesity-related subclinical inflammation have not yet been recognized.
Fecal Microbial Transplant (FMT) has been used to treat some diseases since the 4th century! In this technique, fecal bacteria from healthy donors are administered to recipients who are suffering from a complication (Gupta et al., 2016) [72]. The gut microbiota digests the undigested food, producing SCFA and vitamins; it is effective in gut motility, and it is even effective in mental health status; no surprise alterations made by FMT may exert dramatic effects on the recipient. Currently, disorders such as Inflammatory Bowel Disease (IBD) and recurrent Clostridium Difficile Infection (CDI) can be treated by FMT [73]. Some researchers have employed FMT to cause weight alterations in rats [74]. Metagenomic studies show that people affected by obesity have a higher intestinal bacterial capacity to ferment carbohydrates and produce SCFA. Furthermore, the obese mice and humans have more pathogenic species of Proteobacteria, Bacteroides, Campylobacter and Shigella, and less population of Akkermansia muciniphilae. The latter is an anti-inflammatory species whose reduction will disturb the integrity of the mucosal barrier and increase oxidative stress level [75].
As we saw, manipulating intestinal microbiota seems to have important effects on health status and body weight. However, this is a rather new and open field, calling for more intensive research.
References
- World Health Organization (2018) Obesity and overweight. World Health Organization, Geneva, Switzerland.
- Flegal KM, Kruszon-Moran D, Carroll MD, Fryar CD, Ogden CL (2016) Trends in obesity among adults in the United States, 2005 to 2014. JAMA 315: 2284-2291.
- Segula D (2014) Complications of obesity in adults: a short review of the literature. Malawi Medical Journal 26: 20-24.
- Milaneschi Y, Simmons WK, Rossum EFC, Penninx BWJH (2018) Depression and obesity: evidence of shared biological mechanisms. Molecular Psychiatry 24: 18-33.
- Okamura T, Hashimoto Y, Hamaguchi M, Obora A, Kojima T, et al. (2018) Ectopic fat obesity presents the greatest risk for incident type 2 diabetes: a population-based longitudinal study. International Journal of Obesity 43: 139-148.
- Snel M, Jonker JT, Schoones J, Lamb H, Roos AD, et al. (2012) Ectopic fat and insulin resistance: pathophysiology and effect of diet and lifestyle interventions. International Journal of Endocrinology 2012: 983814.
- Mito N, Hiyoshi T, Hosoda T, Kitada C, Sato K (2002) Effect of obesity and insulin on immunity in non-insulin-dependent diabetes mellitus. European Journal of Clinical Nutrition 56: 347.
- Shulman GI (2000) Cellular mechanisms of insulin resistance. The Journal of Clinical Investigation 106: 171-176.
- Lee B-J, Lin J-S, Lin Y-C, Lin P-T (2015) Antiinflammatory effects of L-carnitine supplementation (1000 mg/d) in coronary artery disease patients. Nutrition 31: 475-479.
- Xu Y, Jiang W, Chen G, Zhu W, Ding W, et al. (2017) L-carnitine treatment of insulin resistance: a systematic review and meta-analysis, Advances in Clinical and Experimental Medicine 26: 333-338.
- Bene J, Hadzsiev K, Melegh B (2018) Role of carnitine and its derivatives in the development and management of type 2 diabetes. Nutrition & Diabetes 8: 8.
- Leggio M, Lombardi M, Caldarone E, Severi P, D’Emidio S, et al. (2017) The relationship between obesity and hypertension: an updated comprehensive overview on vicious twins. Hypertension Research 40: 947.
- Jiang S, Lu W, Zong X, Ruan H, Liu Y (2016) Obesity and hypertension. Experimental and Therapeutic Medicine 12: 2395-2399.
- Laakso M, Kuusisto J (2014) Insulin resistance and hyperglycaemia in cardiovascular disease development. Nature Reviews Endocrinology 10: 293.
- Hammarsten J, Damber J-E, Haghsheno MA, Mellström D, Peeker R (2018) A stage-dependent link between metabolic syndrome components and incident prostate cancer. Nature Reviews Urology 15: 321-333.
- Wang Z, Zou Z, Wang S, Yang Z, Ma J (2018) Chinese famine exposure in infancy and metabolic syndrome in adulthood: results from the China health and retirement longitudinal study. European Journal of Clinical Nutrition 73: 724-732.
- Refaeli R, Chodick G, Haj S, Goren S, Shalev V, et al. (2018) Relationships of H. pylori infection and its related gastroduodenal morbidity with metabolic syndrome: a large cross-sectional study. Scientific Reports 8: 4088.
- Hammarsten J, Peeker R (2011) Urological aspects of the metabolic syndrome. Nature Reviews Urology 8: 483.
- Wong A, Le A, Lee M-H, Lin Y-J, Nguyen P, et al. (2018) Higher risk of hepatocellular carcinoma in Hispanic patients with hepatitis C cirrhosis and metabolic risk factors. Scientific Reports 8: 7164.
- Mohanan S, Tapp H, McWilliams A, Dulin M (2014) Obesity and asthma: pathophysiology and implications for diagnosis and management in primary care. Experimental Biology and Medicine 239: 1531-1540.
- Zhang L, Yin Y, Zhang H, Zhong W, Zhang J (2017) Association of asthma diagnosis with leptin and adiponectin: a systematic review and meta-analysis. Journal of Investigative Medicine 65: 57-64.
- Davis I, Liu A (2015) What is the tryptophan kynurenine pathway and why is it important to neurotherapeutics? Expert Review of Neurotherapeutics 15: 719-21.
- Stone TW, McPherson M, Darlington LG (2018) Obesity and cancer: existing and new hypotheses for a causal connection. EBioMedicine 30: 14-28.
- Deng Y, Zhang Q, Luo H, Chen X, Han Q, et al. (2016) Sustained elevation of NF-κB activity sensitizes offspring of maternal inflammation to hypertension via impairing PGC-1α recovery. Scientific Reports 6: 32642.
- Moustafa JSE-S, Froguel P (2013) From obesity genetics to the future of personalized obesity therapy. Nature Reviews Endocrinology 9: 402.
- Brockmann GA, Tsaih S-W, Neuschl C, Churchill GA, Li R (2009) Genetic factors contributing to obesity and body weight can act through mechanisms affecting muscle weight, fat weight, or both. Physiological Genomics 36: 114-126.
- Herrera BM, Lindgren CM (2010) The genetics of obesity. Current Diabetes Reports 10: 498-505.
- Al-Kloub MI, Froelicher ES (2009) Factors contributing to adolescent obesity. Saudi Medical Journal 30: 737-749.
- Samsell L, Regier M, Walton C, Cottrell L (2014) Importance of android/gynoid fat ratio in predicting metabolic and cardiovascular disease risk in normal weight as well as overweight and obese children. Journal of Obesity 2014: 846578.
- Guerre-Millo M (2002) Adipose tissue hormones. J Endocrinol Invest 25: 855-861.
- Jo J, Gavrilova O, Pack S, JouW, Mullen S, et al. (2009) Hypertrophy and/or hyperplasia: dynamics of adipose tissue growth. PLoSComputBiol 5: e1000324.
- Muir LA, Neeley CK, Meyer KA, Baker NA, Brosius AM, et al. (2016) Adipose tissue fibrosis, hypertrophy, and hyperplasia: correlations with diabetes in human obesity. Obesity (Silver Spring) 24: 597-605.
- Palmer BF, Clegg DJ (2015) The sexual dimorphism of obesity. Mol Cell Endocrinol 402: 113-119.
- Gilsanz V, Hu HH, Kajimura S (2013) Relevance of brown adipose tissue in infancy and adolescence. Pediatr Res 73: 3-9.
- Sanchez-Gurmaches J,Guertin DA (2014) Adipocyte lineages: tracing back the origins of fat. Biochim Biophys Acta 1842: 340-351.
- Zfp423 maintains white adipocyte identity through suppression of the beige cell thermogenic gene program. Cell Metab 23: 1167-1184.
- Raje V (2015) JAK inhibition induces browning of white adipocytes. J Postdoctoral Res 35: 36.
- Sharma BK, Patil M, Satyanarayana A (2014) Negative regulators of Brown Adipose Tissue (BAT)-mediated thermogenesis. J Cell Physiol 229: 1901-1907.
- Coelho M, Oliveira T,Fernandes R (2013) State of the art paper Biochemistry of adipose tissue: an endocrine organ. Arch MedSci 9: 191-200.
- Sender R, Fuchs S, Milo R (2016) Revised estimates for the number of human and bacteria cells in the body. PLoSBiol 14: e1002533.
- Thursby E, Juge N (2017) Introduction to the human gut microbiota. Biochemical Journal 474: 1823-1836.
- Fraher MH, O’toole PW, Quigley EMM (2012) Techniques used to characterize the gut microbiota: A guide for the clinician. Nature Reviews Gastroenterology & Hepatology 9: 312-322.
- Rodríguez JM, Murphy K, Stanton C, Ross RP, Kober OI, et al. (2015) The composition of the gut microbiota throughout life, with an emphasis on early life. Microbial Ecology in Health and Disease 26: 26050.
- Dao MC, Everard A, Clément K, Cani PD (2016) Losing weight for a better health: Role for the gut microbiota. Clinical Nutrition Experimental 6: 39-58.
- Wang J, Tang H, Zhang C, Zhao Y, Derrien M, et al. (2015) Modulation of gut microbiota during probiotic-mediated attenuation of metabolic syndrome in high-fat diet-fed mice. The ISME Journal 9: 1-15.
- Drissi F, Merhej V, Angelakis E, Kaoutari A, Carriere F, et al. (2014) Comparative genomics analysis of Lactobacillus species associated with weight gain or weight protection. Nutrition & Diabetes 4: e109.
- Ohira H, Tsutsui W, Fujioka Y (2017) Are short chain fatty acids in gut microbiota defensive players for inflammation and atherosclerosis? Journal of Atherosclerosis and Thrombosis 24: 660-672.
- Burger-van Paassen N, Vincent A, Puiman PJ, Van Der SM, Bouma J, et al. (2009) The regulation of intestinal mucin MUC2 expression by short-chain fatty acids: implications for epithelial protection. Biochemical Journal 420: 211-219.
- Byrne CS, Chambers ES, Morrison DJ, Frost G (2015) The role of short chain fatty acids in appetite regulation and energy homeostasis. International Journal of Obesity 39: 1331-1338.
- Dodds WJ (2017) Central Nervous System Regulation of Appetite in Humans and Pet Animals. Ann Clin Exp Metabol 2: 1013.
- van de Wouw M, Schellekens H, Dinan TG, Cryan JF (2017) Microbiota-gut-brain axis: modulator of host metabolism and appetite. J Nutr 147: 727-745.
- Triantafilou M, Triantafilou K (2002) Lipopolysaccharide recognition: CD14, TLRs and the LPS-activation cluster. Trends Immunol 23: 301-304.
- Fischer CW, Elfving B, Lund S, Wegener G (2014) 62. Chronic LPS administration induces prolonged sickness behavior in rats. Brain, Behavior, and Immunity 40: e18-e19.
- Wang L, Li L, Ran X, Long M, Zhang M, et al. (2013) Lipopolysaccharides reduce adipogenesis in 3T3-L1 adipocytes through activation of NF-κB pathway and downregulation of AMPK expression. Cardiovasc Toxicol 13: 338-346.
- Zhao M, Chen X (2015) Effect of lipopolysaccharides on adipogenic potential and premature senescence of adipocyte progenitors. Am J Physiol Endocrinol Metab 309: E334-E344.
- Cani PD, Amar J, Iglesias MA, Poggi M, Knauf C, et al. (2007) Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 56: 1761-1772.
- Rorato R, Borges BC, Uchoa ET, Antunes-Rodrigues J, Elias CF, et al. (2017) LPS-induced low-grade inflammation increases hypothalamic JNK expression and causes central insulin resistance irrespective of body weight changes. Int J Mol Sci 18: 1431.
- Cotter PD, Hill C, Ross RP (2005) Food microbiology: bacteriocins: developing innate immunity for food. Nat Rev Microbiol 3: 777-788.
- Saavedra L, Castellano P, Sesma F (2004) Purification of bacteriocins produced by lactic acid bacteria. Methods Mol Biol 268: 331-336.
- Zhang Y, Zhang H (2013) Microbiota associated with type 2 diabetes and its related complications. Food Science and Human Wellness 2: 167-172.
- Mazidi M, Rezaie P, Ferns G, Vatanparast H (2017) Impact of probiotic administration on serum c-reactive protein concentrations: systematic review and meta-analysis of randomized control trials. Nutrients 9: 20.
- Lv Y, Zhao X, Guo W, Gao Y, Yang S, et al. (2018) The relationship between frequently used glucose-lowering agents and gut microbiota in type 2 diabetes mellitus. J Diabetes Res 2018:1890978.
- Gu Y, Wang X, Li J, Zhang Y, Zhong H, et al. (2017) Analyses of gut microbiota and plasma bile acids enable stratification of patients for antidiabetic treatment. Nat Commun 8: 1785.
- Kitai T, Tang WHW (2018) Gut microbiota in cardiovascular disease and heart failure. Clin Sci (Lond) 132: 85-91.
- Tang WHW, Kitai T, Hazen SL (2017) Gut microbiota in cardiovascular health and disease. Circ Res 120: 1183-1196.
- Cho Y, Shore SA (2016) Obesity, asthma, and the microbiome. Physiology 31: 108-116.
- Shore SA, Cho Y (2016) Obesity and asthma: microbiome–metabolome interactions. Am J Respir Cell Mol Biol 54: 609-617.
- Rogers CJ, Prabhu KS, Vijay-Kumar M (2014) The microbiome and obesity-an established risk for certain types of cancer. The Cancer Journal 20: 176-180.
- Djuric Z (2017) Obesity-associated cancer risk: The role of intestinal microbiota in the etiology of the host proinflammatory state. Translational Research 179: 155-167.
- Cani PD, Jordan BF (2018) Gut microbiota-mediated inflammation in obesity: A link with gastrointestinal cancer. Nature Reviews Gastroenterology & Hepatology 15: 671-682.
- Govan JRW (1986) In vivo significance of bacteriocins and bacteriocin receptors. Scandinavian Journal of Infectious Diseases 49: 31-37.
- Gupta S, Allen-Vercoe E, Petrof EO (2016) Fecal microbiota transplantation: in perspective. Therapeutic Advances in Gastroenterology 9: 229-239.
- Khanna S, Vazquez-Baeza Y, González A, Weiss S, Schmidt B, et al. (2017) Changes in microbial ecology after fecal microbiota transplantation for recurrent C. difficile infection affected by underlying inflammatory bowel disease. Microbiome 5: 55.
- Kang Y, Cai Y (2017) Gut microbiota and obesity: implications for fecal microbiota transplantation therapy. Hormones 13: 223-234.
- Valsecchi C, Carlotta ST, Castellazzi A (2016) Gut microbiota and obesity. Journal of Clinical Gastroenterology 50: 157-158
Citation: Taghizad F (2021) The Roles of Bacterial Products, Lipopolysaccharides and Bacteriocins, in Obesity and its Related Complications. J Diabetes Metab Disord 8: 039.
Copyright: © 2021 Fereidoun Taghizad, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.