The Structural Hybrids of Acetylcholinesterase Inhibitors in the Treatment of Alzheimer’s Disease: A Review
*Corresponding Author(s):
Anil K SaxenaDivision Of Medicinal And Process Chemistry, CSIR Central Drug Research Institute, Lucknow, India
Tel:+91 9839012951,
Email:anilsak@gmail.com
Abstract
Alzheimer’s Disease (AD) is characterized by the loss of memory and learning ability in elderly patients affecting large population worldwide. The enzyme Acetylcholinesterase (AChE), (E.C.3.1.1.7) plays a major role in the hydrolysis of the released neurotransmitter acetylcholine. Most of the clinically used drugs to treat AD are Acetylcholinesterase Inhibitors (AChEIs). These drugs can provide symptomatic benefits only and suffer with loss of therapeutic potential with time. Therefore, there is an urgent need of novel cholinesterase inhibitors with wider therapeutic window for the treatment of AD. The strategies targeting the AChE enzyme along with other target(s) like Butyrylcholinesterase (BChE), amyloid-β (Aβ), β-secretase-1 (BACE), metals (Cu2+, Zn2+, or Fe2+), antioxidant properties and free radical scavenging capacity have been mainly focused in the last five years. A number of hybrid molecules incorporating sub-structures with the desired well-established pharmacological profile into a single scaffold have been investigated. The main sub structures used in developing these molecules are derived from diverse chemical classes such as acridine, quinoline, carbamates, huperzine and other heterocyclic analogs. It has been followed by optimization of activity through structural modifications of the prototype molecules for developing the Structure Activity Relationship (SAR). This has led to the development of novel molecules with desired AChE inhibitory activity along with other desirable pharmacological properties. This review summarizes the current therapeutic strategies for the development of these AChEI in the last seven years.
Keywords
ABBREVIATIONS
AD - Alzheimer’s Disease
ACh - Acetylcholine
AChE - Acetylcholinesterase
BChE - Butyrylcholinesterase
Aβ - Amyloid Beta
CAS - Catalytic Anionic Site
PAS - Peripheral Anionic Site
CNS - Central Nervous System
THA - Tetrahydroacridine
DPPH - 1,1-diphenyl-2-picrylhydrazyl
ABTS - (2,20-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid)
IC50 - The half maximal Inhibitory Concentration
APP - Amyloid Precursor Protein
INTRODUCTION
Alzheimer’s Disease (AD) is the most common form of irreversible neurological disorder of elderly patients that has affected large population worldwide [1]. It is clinically characterized by progressive cognitive impairments or defined by a loss of memory and learning ability, together with a reduced ability to perform daily routine activities and adverse array of neuropsychiatric symptoms such as apathy, verbal and physical agitation, irritability, anxiety, depression, delusions and hallucinations [2]. Characteristic neuropathologic findings include selective neuronal and synaptic losses, extracellular neuritic plaques containing the β-amyloid peptide and Neurofibrillary Tangles (NFTs) composed of hyperphosphorylated forms of the tau (τ) protein [3]. Still existing treatments for mild to moderate AD include the use of Acetylcholinesterase (AChE) inhibitors such as tacrine, donepezil ivastigmine, galanthamine, (Figure1) and the use of N-methyl-D-aspartate (NMDA) antagonist memantine [4]. However, these drugs are unable to slow or prevent AD progression, rather can provide symptomatic benefits only and suffer from major drawback of loss of therapeutic potential with time. Thus, increasing daily doses in such circumstances increases the side effects until the pause of the treatment. The major side effects are specifically caused by the peripheral activity of these drugs on cholinesterase enzyme. As the average age is increasing all over the world, and so the AD (66% in the developing countries), there is an urgent need for novel therapeutics, which could act as anti-Alzheimer agents with less side effects than the known commercial drugs for the treatment of the AD. The clinical trial results from current developmental pipeline [5] involving molecules as anti-amyloid, anti-tau, neuroprotective (calpain inhibitor, estrogen receptor beta agonist, Peroxisome Proliferator-Activated Receptor (PPAR)-gamma agonist, Gamma-Aminobutyric Acid (GABA)-receptor modulator), Phosphodiesterase (PDE)-9A inhibitor, Butyrylcholinesterase (BChE) inhibitor, 5-Hydroxytryptamine (5-HT)-6 receptor antagonist etc., but the success rate is very poor (0.4%) due to high attrition rate, 99.6% indicate that the most of the candidate drugs are failing to meet the primary cognitive and functional end points [5-7]. Hence extensive efforts are made for safe and effective anti-Alzheimer’s agents [8]. Among several targets explored, the AChE is the major target in providing clinically effective anti-Alzheimer’s agents.
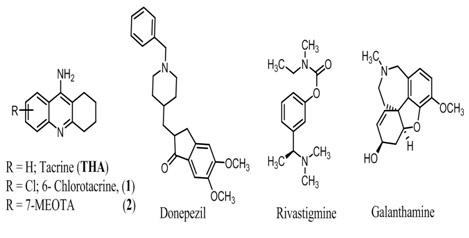 Figure 1: Structures of tacrine and it analogs, donepezil, rivastigmine and galanthamine.
Figure 1: Structures of tacrine and it analogs, donepezil, rivastigmine and galanthamine.
CHOLINERGIC HYPOTHESIS
In search for novel therapeutic approaches towards AD, the mechanism-based therapeutic approaches targeting β-amyloid, tau pathologies and cholinergic hypothesis have been mainly addressed in last several years [3]. The cholinergic hypothesis [9] has been the most widely used in search of drug(s) for treatment of AD. According to this hypothesis, enzyme AChE hydrolyzes the neurotransmitter ACh and breakdown it into acetic acid and choline (Figure 2) and creates deficiency of the ACh at the synaptic gap which leads to the termination of synaptic transmission in brain. The AD is caused by reduced level of the neurotransmitter Acetylcholine (ACh) at the synaptic gap in central nerve system in mammals. There are two cholinesterase enzymes that metabolize acetylcholine, namely, a) AChE and b) BChE. The AChE is mainly found in neuromuscular junctions and chemical synapses of the cholinergic type and plays a major role in the hydrolysis of the released neurotransmitter ACh in central nervous system.
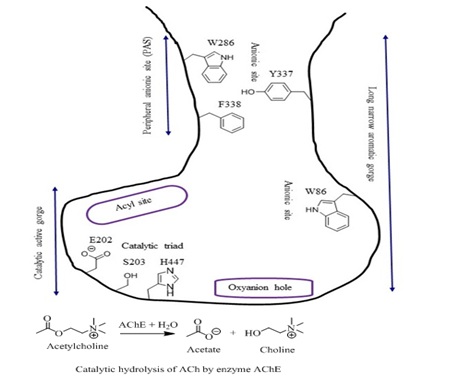 Figure 2: Diagrammatic view of AChE enzyme active site and catalytic hydrolysis of ACh.
Figure 2: Diagrammatic view of AChE enzyme active site and catalytic hydrolysis of ACh.
STRUCTURE OF ACETYLCHOLINESTERASE
X-ray crystallography of AChE enzyme in Torpedo californica (eeAChE) co-crystalized with tarcrine has shown that it is a long and narrow gorge lined by 14 amino acid residues. The gorge possesses two separate ligand binding sites, one is Catalytic Active Site (CAS) located at the bottom of the gorge and the second is Peripheral Anionic Site (PAS) which is about 15 Å above the catalytic active site and located at the entrance of the gorge [10].
According to the recently reported the crystal structures of human AChE (hAChE) in complex with Fasciculin 2 (FAS-2) and huprine W (PDB code 4BDT) the catalytic active site is lined by aromatic residues and itself can be divided into two sub-unites namely, catalytic triad (also known as esteratic sub-site) and anionic sub-site. The catalytic triad sub-site consists glutamic acid (E202), serine (S203) and histidine (H447), while anionic sub-site consists tryptophan (W86). The catalytic triad is involved in the hydrolysis of the neurotransmitter acetylcholine, while the quaternary group of the choline moiety interacts with of tryptophan (W86) of the anionic site of the CAS. The adjacent area of catalytic triad is known as “oxyanion hole” which is formed by glycine and alanine residues and interacts with carbonyl oxygen of ACh. Whereas the PAS consisting of tryptophan (W286), tyrosine (Y337) and phenylalanine (F338) is believed to guide the entrance of suitable choline esters into the catalytic domain of AChE [11] (Figure 2). The binding of any ligand at PAS blocks the entry of substrates and the exit of the products from the base of the active site. Some ligands such as bulky bis quaternary compounds bind with PAS and hence block the entrance of acetylcholine in the catalytic gorge therefore partially inhibit activity of AChE.
SELECTIVITY FOR ACHE OVER BCHE
It is evident that AChE promotes amyloid fibril formation, more specifically; the peripheral site of enzyme AChE actively involved in aggregation of amyloid-beta (Aβ) peptides [12]. The AChE interacts with growing amyloid fibrils and form AChE-Amyloid complex that accelerates assembly of Aβ peptides which leads to Aβ deposits such as mature senile plaque present in the AD brain [13,14]. The AChEIs bind only with CAS prevents the hydrolysis of ACh thus rectify the deficit of ACh in brain. Whereas the AChEI which blocks the entire active gorge including catalytic active site and peripheral site of enzyme of AChE, known as dual binding site inhibitors not only prevent the hydrolysis of ACh but also inhibit Aβ aggregation. So these dual AChEIs are considered more effective anti-Alzheimer’s agents than the single biding site inhibitors.
The BChE is produced in liver and is mainly found in blood plasma and plays secondary role in hydrolysis of ACh while AChE found in central nerve system plays a lead role in ACh hydrolysis. Recent research indicated that BChE prevent Aβ-fibril formation and hence delay the formation of Aβ-aggregation in brain which is contrary to AChE [15,16]. Therefore, high selectivity of the ligand toward AChE over BChE is highly desired property to treat AD. The BChE possesses some structural similarities at CAS but lacks PAS and it lacks three of four aromatic residues of the AChE PAS. This distinct feature may be used in designing selective AChEIs. The hybrid AChEIs are derived by molecular hybridization of two structural fragments that are well known for their pharmacological profile. These novel hybrid AChEIs incorporating additional pharmacophores for binding at PAS may also offer selectivity over BChE. This review compiling these hybrid AChEIs may provide detailed insight into the design and optimization through Structure-Activity-Relationship (SAR) studies in the search of novel candidate molecules.
IRREVERSIBLE, QUASI-IRREVERSIBLE AND REVERSIBLE ACHE INHIBITORS
The AChEIs can be divided into three categories, irreversible, quasi-irreversible and reversible based on the mechanism of their interaction with AChE. The irreversible inhibitors covalently bind with serine (S202) of catalytic triad in CAS and hence block the active site permanently, e.g., organ phosphorous. The irreversible AChEIs are used as insecticides pesticides and warfare which is out of the scope of the present review. The reversible and quasi-irreversible inhibitors have two key interactions such as π-cation interaction with tryptophan (W86) and hydrogen bonds with serine (S202) of active gorge of AChE enzyme in huprine W-AChE complex [11]. These types of inhibitors have more therapeutic applications as anti-alzheimer’s agents due to their ability to modulate ACh to appropriate levels. Thus these inhibitors are discussed in this review. They are mainly of three types based on their pharmacokinetic mechanism, i) competitive, ii) non-competitive and iii) mixed type inhibitors. The detailed studies of pharmacokinetic mechanism are out of the scope of this review article. However, the necessary and relevant information of binding mechanism of AChEIs is discussed at appropriate places in this review.
In search of anti-Alzheimer’s drugs, intensive efforts have been focused on the AChE target, because of the importance of cholinergic hypothesis in the development of clinically effective anti-Alzheimer’s agents. It resulted in the successful launch of 4 drugs specifically AChE inhibitors such as tacrine (tetra hydro amino acridine or THA) donepezil, rivastigmine, and galanthamine (Figure 1) [4]. The tacrine, (Figure 1) was withdrawn from the market due to serious hepatotoxicity [17]. The less toxic drugs, donepezil, inhibit only AChE while rivastigmine acts against both enzymes AChE as well as BChE, exhibiting thus a higher efficiency. Overall, these drugs show modest improvement in the cognitive function of alzheimer’s disease, and suffer from major drawback of loss of therapeutic potential with time.
HYBRID STRATEGY
In search of safer and more potent anti-Alzheimer’s agents than the above existing drugs particularly acting as AChEIs different chemical classes have been investigated and reviewed in last five year (2011-2015) [18-22]. Researchers developed a series of innovative hybrid AChEIs analogs by conjoining various active scaffolds with existing drug molecules. These analogs provided promising leads for the development of anti-Alzheimer’s agents. The active scaffold in these analogs included coumarin, flavonoid, phenothiazine, benzothiazole, aryl, pyridine, indole, isoindoline-1,3-dione, melatonin, thiadiazolidinone, β-carboline, ferulic or caffeic acid, thioacetyl, acridine- selegiline, acridine-aminothiazol and piperazine. These hybrid structures exhibited much higher AChE inhibitory activities as compared to their non-hybrid precursors [23,24]. In addition, these hybrids acted as multifunctional agents, such as anti-oxidant, metal ion chelator, neuroprotective, anti-Aβ aggregation and anti-inflammatory agents in the treatment of AD and are discussed below.
In this review we have summarized the systematic development of key AChEIs claimed in research papers and major reviews published in last five years (2011-2015). The potent AChEIs with diverse chemical structures including, hybrid multi target, dual binding site, homo/heterodimers, heterocyclic, small molecules and natural products have been analyzed under broad chemical classes from medicinal chemistry point of view.
TACRINE ANALOGS
This chemical class gave the first clinically useful drug tacrine (Figure 1) for treating Alzheimer’s disease. However, Watkins, reported it’s some serious toxicity [17], its 6-chloro analogs (Figure 1) showed stronger AChE binding than tacrine [25]. The 7-methoxytetrahydroacridine (7-MEOTA, 2, Figure 1) was weaker cholinesterase inhibitor in human (hAChE IC50 = 15000 nM, BChE IC50 = 21000 nM) than tacrine (AChE IC50 = 500 nM) in vitro and in vivo studies [26,27]. Most importantly, its lower toxicity and better antioxidant properties, gave advantages over tacrine therefore should be consider for further development. Later on researchers found that the catalytic site of AChE is involved in the hydrolysis of the neurotransmitter acetylcholine, whereas the peripheral site is involved in a binding process with Amyloid-β (Aβ) that accelerates the aggregation of this peptide [12]. Thus, simultaneous blockage of the catalytic and peripheral anionic sites of AChE by dual binding site AChEIs resulted in the simultaneous modulation of two important targets of AD pathology, namely Aβ aggregation and the cholinergic deficit [18,19,22]. This strategy led to the development of homo/heterodimer or hybrids of two moieties known for their anti-alzheimer properties and such tacrine analogs are discussed below [28,29].
A series of bis-tacrines homodimers linked with 6-8 methylene units showed potent inhibition towards AChE [30]. Among these heptylene-linked bis-tacrineor bis(7)-tacrine (B7T, 3a in Figure 3 and Table 1) was 1,000-fold more potent than tacrine in inhibiting rat brain rAChE (IC50 = 0.2 nM) and rat serum BChE (IC50 = 315 nM) with high selectivity index (SI = 1369.6) towards AChE over BChE, indicating that it is more selective and effective AChE inhibitor than tacrine (rAChE, IC50 = 250 nM, rBChE IC50= 40 nM, SI = 0.2). This may be due to its interaction with both catalytic and peripheral anionic sites. These dual site binding AChE inhibitors led to a new therapeutic option which later culminated in designing of a large number of dual-site binding AChEIs. Using this strategy, a huge number AChEIs have been extensively coupled with known pharmacophore entities with well-established biological activities, to develop homo- and heterodimers with improved pharmacological profile. In the recent past, researchers developed series of innovative acridine-homo/heterodimer analogs by conjoining various heterocyclic moieties with tacrine [28-33]. These analogs have been helpful in providing promising leads for the development of anti-alzheimer’s agents [28,29,31-34]. In 2012, Hamulakova et al., reported a series of tacrine homodimers (4a-b in Figure 3 and Table 1) linked through a piperazine/thiourea linker, among them the bis tacrine analog 4a having piperazine linker, displayed 111 fold more hAChE (IC50 = 4.49 nM) inhibitory activity than its parent drug tacrine (IC50 = 500 nM) and showed 338.5 times more selectivity towards AChE over BChE (IC50 = 1520 nM) [35]. While the other bis tacrine analog 4b having ethylene-thiourea linker showed 250 fold more hAChE (IC50 = 2 nM) inhibitory activity than tacrine (IC50 = 500 nM) with poor selectivity (SI = 0.25) over BChE (IC50 = 8 nM) [36]. These results indicated that piperazine linker was better choice to achieve high selectivity. Recently, a series of bistacrine homodimers (5a-f of Figure 3) with various linkers (alkyl, ether, amine or amide) between two tacrine moieties, were investigated for their pharmacological, pharmacokinetic and physicochemical properties [37]. Among them the 5c showed highest AChE inhibitory activity (IC50 = 1.1 nM) and 213 fold selectivity over BChE (IC50 = 234 nM). All the tested dimers showed significantly lower cytotoxicity, intestinal permeability than that of tacrine. It was observed that replacement of tetra hydro ring of tacrine moiety dramatically decreased the AChE inhibitory activity.
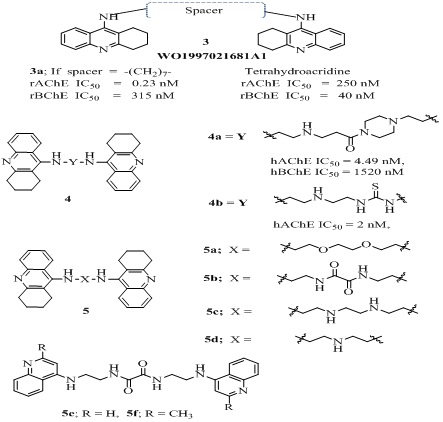 Figure 3: Structures of tacrine-homodimers.
Figure 3: Structures of tacrine-homodimers.
|
Compound No. |
Screening model |
Activity IC50 nM |
Compound No. |
Screening model |
Activity IC50 nM |
Compound No. |
Screening model |
Activity IC50 nM |
|
3a |
rAChE |
0.23 |
14 |
AChE |
10.9 |
23 |
eeAChEp |
5 |
|
Tecrine |
rAChE |
250 |
15 |
eeAChE |
9.1 |
24 |
hAChE |
470 |
|
4a |
hAChE |
4.49 |
16 |
rAChE |
5.87 |
25 |
hAChE |
1350 |
|
4b |
hAChE |
2 |
17 |
eeAChE |
2.37 |
26 |
eeAChEp |
22.6 |
|
6a |
eeAChE |
7.98 |
18a |
hAChE |
0.035 |
27 |
hAChE |
1.18 |
|
6b |
eeAChE |
4.55 |
18b |
hAChE |
8 |
28 |
hAChE |
0.006 |
|
8a |
eeAChE |
308 |
18c |
hAChE |
12.8 |
29 |
eeAChE |
37 % inhibition at 10mg/kg |
|
8b |
eeAChE |
3.65 |
19 |
eeAChE |
133 |
30 |
eeAChE |
46 % inhibition at 10mg/kg |
|
11 |
eeAChE |
17 |
20a |
eeAChE |
92 |
31 |
AChE |
89 |
|
12 |
AChE |
340 |
20b |
eeAChE |
33.63 |
32 |
AChE |
5000 |
|
13 |
eeAChEp |
7.37 |
21 |
hAChE |
15.4 |
33 |
AChE |
63.2 |
|
34 |
hAChE |
0.72 |
38 |
AChE |
7300 |
43 |
AChE |
768 |
|
35a |
hAChE |
142 |
39a |
hAChE |
80 |
44 |
AChE |
45 |
|
35b |
AChE |
19 |
40 |
AChE |
40 |
45 |
AChE |
270 |
|
36 |
hAChE |
0.2 |
41 |
AChE |
17 |
46 |
eeAChE |
69 |
|
37 |
hAChE |
22.2 |
42 |
hAChE |
780 |
47 |
eeAChE |
25 |
Table 1: The screening model and activity IC50 of Tacrine Analogs.
Among the tacrine-multi alkoxybenzene hybrids (6 of Figure 4, Table 1) derived from conjoin of acridine and substituted aryl group through amine/amide linkers of appropriate length, the most active hybrid (6a of figure 4) inhibited both AChE in Electric eel (eeAChE, IC50 = 7.98 nM) and BChE in equine serum (esBChE, IC50 = 7.94 nM) equally with no selectivity and also showed higher self-induced Aβ aggregation inhibition compare to curcumin [38]. Later, the same research group reported improved version of these hybrids (6b of Figure 4) with slightly better eeAChE (IC50 = 4.55 nM) and esBChE (IC50 = 3.41 nM) activities [39]. These hybrids containing hydroxyl substituents, additionally exhibited significant antioxidant activity and suppressed self-induced Aβ aggregation. The hydrochloride salt of the analog 7 (Figure 4) obtained by joining tacrine with 4-fluorobenzoic acid through an alkyl chain linker, was 4 fold more effective AChEIs (IC50 = 0.784 nM) than tacrine. However, it was more selective towards BChE (IC50= 0.0354 nM) over AChE [40]. Among the analogs 8 (Figure 4, Table 1) reported as hybrids of tacrine and hydrazine nicotinate moiety by the same research group [41,42], the analog 8a was weaker eeAChE inhibitor (IC50 = 308 nM) than the reference tacrine (IC50 = 5.46 nM) and was selective towards esBChE (IC50 = 0.65 nM) [41]. The other analog 8b, with longer alkyl spacer and smaller size of tricyclic ring compared to 8a, was stronger AChEI (IC50 = 3.65 nM) than tacrine (IC50 = 5.46 nM) with 4685 times selectivity towards AChE over BChE (IC50 = 17100 nM) [42]. The analogs 9, 10 (Figure 4) having thienopyridine joined with phthalimide and lipoic acid moieties respectively through a linker, showed 56% and 57% AChE enzyme inhibition respectively at 10mg/kg oral dose in adult male albino Wister rats similar to tacrine (55% AChE inhibition) [43]. A series of tacrine-phenyl-benzothiazole hybrids (11, 12 of Figure 5) were derived by connecting tacrine with benzothiazole moiety through an alkyl/amide spacer [44]. Among them the most potent hybrid (11 of Figure 5) was 18 times more effective in eeAChE inhibition (IC50 = 17 nM) than tacrine (IC50 = 311 nM) and 7 times more selective towards AChE than esBChE (IC50 = 122 nM). Moreover, it exhibited equally Aβ self-aggregation inhibition (IC50 = 51.8%) as its reference curcumin (IC50 = 52.1%) at 20 µM concentration. The other tacrine-benzothiazine analog 12 exhibited good AChE inhibition (IC50 = 340 nM) comparable to that of tacrine (IC50 = 190 nM) [45]. More importantly it showed self-mediated Aβ42 aggregation inhibition (22.3% at 50 µM), poor antioxidant activity (32% quenching at 1.32 mM) in 2, 2-diphenyl-1-picrylhydrazyl (DPPH) free radical assay. However, it’s high lipophilic character and the low Blood-Brain Barrier (BBB) permeability limited drug eligibility. The tacrine-mercapto derivatives 13 (Figure 5) were investigated and among these analog the most potent analog 13 possessed interesting pharmacological profile with eeAChE (IC50 = 42.6 nM) and esBChE (IC50= 91 nM) inhibitory activity, neuroprotective effect (34.0% at 30 µM) in human neuroblastoma cell line F against H2O2 and less hepatotoxicity [46]. These results indicated that tacrine-mercapto derivatives were safer than tacrine, however the high selectivity towards BChE than AChE need attention. The tacrine–ferulic acid–nitrate hybrids [47] were found to be highly potent analogs. The representative analog (14 of Figure 5, Table 1) of the series, showed high in vitro cholinesterase inhibition (AChE IC50 = 10.9 nM, BChE IC50 = 17.7 nM), cognition improving potency and less hepatotoxicity than tacrine. Based on these findings the same research group later, reported the tacrine–flurbiprofen–nitrate hybrid (15 of figure 5) [48] with higher cholinesterase inhibitory activity (AChE IC50 = 9.1, BChE IC50 = 2.5 nM) and impressive pharmacology and pharmacokinetic profile with significant Aβ inhibitory activity and lesser hepatotoxicity than tacrine. Furthermore, it improved the memory impairment caused by scopolamine-induction (passive avoidance test) in adult mice and exhibited moderate blood vessel relaxative activity by releasing Nitric Oxide (NO), indicative of promising lead compound with high safety. The tacrine-lophine hybrid [49] (16 of Figure 5) was designed by connecting tacrine with lophine moiety through an octane linker. It possessed rAChE (IC50 = 5.87 nM) inhibition comparable to bis (7) tacrine (IC50 = 4.12 nM) with high selectivity for AChE over BChE (IC50 = 109 nM, SI = 18).
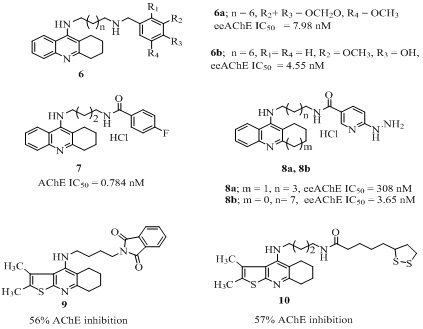 Figure 4: Structures of various tacrine heterodimers.
Figure 4: Structures of various tacrine heterodimers.
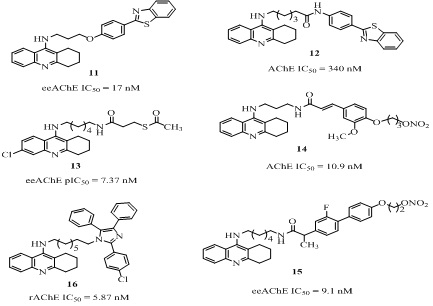 Figure 5: Tacrine analogs with their AChE inhibition (IC50) values.
Figure 5: Tacrine analogs with their AChE inhibition (IC50) values.
The hybrid 17 (Figure 6) 6-chlorotacrine-N1, N1-dimethyl-N4-(pyridin-2-ylmethyl) benzene-1,4-diamine was displayed significant ChE inhibition, Aβ-modulation, metal chelation, BBB permeability [50]. More particularly, it inhibited eeAChE (IC50 = 2.37 nM), where 6-chloroacrine part may be responsible for binding with catalytic site of the enzyme and N1,N1-dimethyl-N4-(pyridin-2-ylmethyl) benzene-1,4-diamine part may be responsible for metal free, metal induced Aβ-modulation and diamino-octyl linker chelated with metal ions (Cu+2, Zn+2). It was also observed that presence of metal ions and Aβ-aggregate assembly did not affect the ChE inhibition ability. Additionally, it has good BBB penetrability and may be considered as a promising multifunctional lead compound. A series of tacrine-flavonoid hybrids (18 of Figure 6, Table 1) was reported for its effective AChE, BChE, BACE-1 inhibition, Oxygen-Radical Absorbance Capacity (ORAC) and in vitro Central Nervous System (CNS) penetration [51]. Among them the most effective analog 18a with hAChE inhibition (IC50 = 0.035 nM) was 10,000 fold better than tacrine and was 143 times more selective for hAChE over hBChE (IC50 = 5.0 nM). The strong AChE inhibitory activity and selectivity of these analogs may be because of better fitting of flavonoid moiety in the PAS of AChE enzyme. In addition, this analog 18a also showed effective antioxidant activity (0.3 trolox equiv.) in oxygen radical absorbance capacity by fluorescence (ORAC-FL) method and high in vitro permeability in BBB (8.7Pe(µM/S) measured in Parallel Artificial Membrane Permeation (PAMPA) assay for BBB (PAMPA−BBB). However, the analog 18b (Figure 6) showed better overall drug profile with balanced combination of hAChE (IC50 = 8.0 nM) and hBChE (IC50 = 1.5 nM) inhibition, ORAC (1.3trolox equiv.), BBB penetration (23.1, Pe(µM/S) for PAMPA−BBB assay) and human recombinant BACE-1 (IC50 = 2800 nM) protein inhibitory activity than the analog 18a [51]. Another tacrine-flavonoid hybrids 18c (Figure 6) having 5,6,7-trimethoxy substituted chromone moiety, displayed 6 fold higher eeAChE (IC50 = 12.8 nM) than tacrine (IC50 = 78.5 nM) with 36 fold selectivity towards AChE [52]. The effective Aβ aggregation inhibition (33.8% at 25 µM), antioxidant activity (0.55 trolox equiv. in ORAC), protective effect against the neurotoxicity induced by H2O2 (45.3% at 5µM) in PC12 cell injury was evaluated via 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium (MTT) assay and BBB permeability in vitro explained its potency as a multi-targeted lead compound. The tacrine-flavonoid 19 (Figure 6) where tacrine and flavonoid moiety were connected through a piperazin linker, moderately inhibited ChE (eeAChE IC50 = 133, esBChE IC50 = 558 nM respectively) [53]. Moreover, it also showed significant metal chelating (Cu2+and Fe2+), inhibition of ChE and self-induced Aβ aggregation and low neuroblastoma cell toxicity in MTT assay. A series of novel tacrine-coumarin heterodimers were designed, synthesized and biologically evaluated for their potential cholinesterase inhibitory activity. Among them the tacrine-coumarin heterodimer 20a (Figure 6) where tacrine was connected with 7-position of coumarin moiety through piperazine spacer, displayed the significant ability to inhibit ChE (AChE, BChE, IC50 = 92, 234 nM respectively). Additionally it also showed a good Aβ(1-42) aggregation inhibition (67.8%, at 20 µM), metal (Cu+2, Fe+2) chelation and non-toxicity towards SH-SY5Y cells [54]. The other tacrine-coumarin analog of the series 20 b (Figure 6) having 3,4-dimethyl substituted coumarin moiety, exhibited better eeACh inhibition (IC50 = 33.63 nM, esBChE IC50= 80.72 nM) than the analog 20a but with low selectivity (SI = 7.6) [55]. Moreover, this analog 20b exhibited selective Monoamine Oxidase-B (MAO-B) inhibition (IC50 = 240 nM), which was approximately 32 times higher than the reference iproniazid (IC50 = 7590 nM) with low toxicity and good in vitro BBB permeability (Pe = 4.75µM/S). It was a competitive inhibitor and acted through mixed-type mechanism with binding affinity (Ki = 34.4 nM) towards eeAChE and molecular modeling studies indicated that it was potential multi-target lead compound. The reported tacrine-coumarin heterodimer 21(Figure 6) where tacrine was connected with 4-position of coumarin, showed excellent hChE inhibition (AChE IC50 = 15.4 nM, BChE IC50= 328 nM) which was approximately 32 times higher than tacrine with high selectivity index for AChE (SI = 21.3) [56]. The high dissociation constant (Ki = 4.1 nM) for AChE-inhibitor complex indicated its strong binding ability with hAChE which was 55 times higher than tacrine. The next hybrid 22 (Figure 6) where tacrine was connected with coumarin moiety at 3-position through an amide linker, displayed higher binding free energy (ΔGcal = -12.7 kcal/mol) than tacrine (ΔGcal = -11.9 kcal/mol) in AChE. Its high dissociation constant (Ki =16.7 nM) of AChE-inhibitor complex, explained its higher potency over tacrine [57]. The molecular modeling, simulation and kinetic studies of the above mentioned tacrine-coumarin analogs indicated that the potency of the compound largely depends on binding mode of the hybrid with the enzyme AChE. The proper orientation of tacrine moiety in CAS, coumarin moiety in the PAS of the gorge and linker of appropriate length having nitrogen atom are key features of the active compounds.
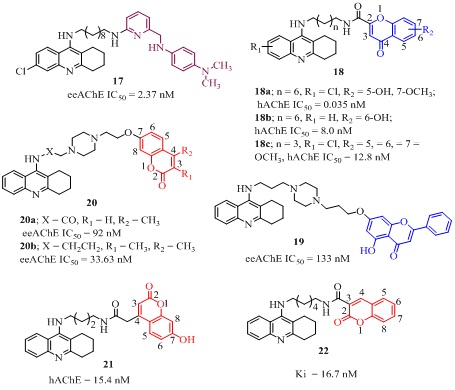
Figure 6: Structures of tacrine analog with their IC50 values.
The other type of tacrine-coumarin hybrid [58] (23 of Figure 7) showed potent eeAChE (IC50 = 5 nM) and esBChE (IC50 = 15700 nM) with 3149 fold selectivity over BChE. The AChE inhibitory activity decreased with substitution at 4-position in the order F>CH3=OCH3=H>Cl, indicating that the electron withdrawing group at 2 and 4 position increases the activity. The heterodimer of 7-MEOTA and NMDA antagonist 1-adamantylamin (24 of Figure 7, Table 1) connected through a thiourea linker, inhibited AChE (IC50 = 470 nM) and BChE (IC50 = 110 nM) moderately with poor selectivity [59]. The other analog of the series 7-MEOTA-p-anisidine hybrid (25 of Figure 7) showed weaker AChE inhibition (IC50= 1350 nM) than tacrine (IC50 = 320 nM) but stronger than 7-MEOTA (IC50 = 10,000 nM) [60]. Among the reported tacrine-selegiline heterodimers [61], the analog 26 (Figure 7) showed significant AChE (IC50 = 22.6 nM), BChE (IC50 = 9.39 nM) and MAO-A/B (0.37, 0.18 µM) inhibition [62]. The tacrine-melatonin heterodimer [62] 27 (Figure 7) designed as a hybrid of well-known antioxidant melatonin pharmacophore and AChE inhibitor tacrine, exhibited 378-fold higher AChE inhibition (IC50=1.18 nM) than tacrine and poor selectivity for AChE (BChE, IC50 = 0.28 nM, SI = 0.237). The 1,2,3,4-tetrahydrobenzo [h] [1,6] naphthyridine-6-chlorotacrine hybrids were designed by molecular hybridization of tacrine and naphthyridine moiety [63]. Among them the optimized lead compound 28 (Figure 7) was an excellent AChEI (IC50 = 0.006 nM) with 20,000 fold more selectivity for AChE (BChE, IC50 = 120 nM). It also showed good anti-Aβ(1-42) aggregation activity (52.5% at 10 µM) in E. coli., along with good tau protein aggregation inhibition (40.7 % at 10 µM) and BBB permeability (Pe = 10.5 µM/S) in CNS. Among the benzopyridines-phthalimide analogs, the 29 (Figure 7) exhibited better eeAChE inhibition (36.99%) than tacrine (28.74%) at 10mg/kg [64]. The 3D molecular modeling revealed that benzopyridines fragment binds with catalytic active site while phthalimide fragment binds with peripheral active site of the enzyme. The thienopyridines-phthalimide analog [64] (30 of Figure 7) showed 1.6 fold higher AChE inhibition (46.25%) than tacrine. The tacrine-phenothiazinehetero dimer 31(Figure 8) designed by combining structural features of tacrine and phenothiazine, showed excellent AChE inhibition (IC50 = 89 nM) in rat brain [65,66]. Additionally, it significantly inhibited tau hyper phosphorylation (34.7% at 1 µM) induced by okadaic acid in N2 cell and also interacted with Fibril Beta Amyloid (fAβ(1–40)) using Surface Plasmon Resonance (SPR). It also showed good calculated physiochemical descriptors for intestinal absorption (97.3%) in humans, in vivo skin permeability (Log Kp 2.15cm/h), in vitro plasma protein binding (86.74%) and in vivo BBB penetration (2.88 C. brain/C. blood).
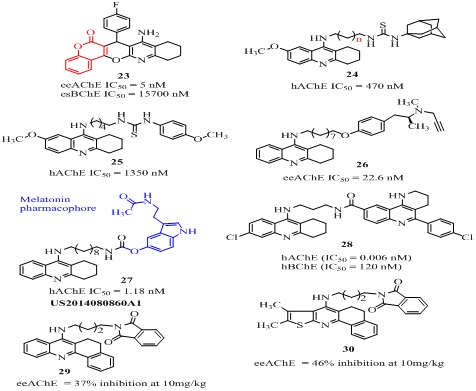 Figure 7: Structures of AChE inhibitors tacrine-hybrids.
Figure 7: Structures of AChE inhibitors tacrine-hybrids.
The tacrine analog 32 (Figure 8) having a nitroxide substituent at 9-amino group via piperazine linker displayed very good AChE inhibitory activity (IC50β-induced cytotoxicity (PC50 < 2000 nM) in MC65 cells [67]. The tacrine-(β-carboline) heterodimers [68] (33 of Figure 8) were evaluated for their role as multifunctional cholinesterase inhibitors. Among them the analog 33 (Figure 8) showed the effective hAChE inhibitory activity (IC50 = 63.2 nM) which was nearly 7-times higher than tacrine (IC50 = 467 nM) with low selectivity over BChE (IC50 = 39.8 nM). It also showed strong inhibition against Aβ aggregation (65.8% at 20 µM), good antioxidant activity (1.57 trolox equiv.), metal ions chelation (Cu+2) and BBB permeability, indicating that it may be a potent multifunctional agent for AD treatment. The tacrine-quinone hybrid 34 (Figure 8) exhibited strong hAChE inhibition (IC50 = 0.72 nM) which was 752 fold more selectivity over BChE (IC50 = 542 nM), along with excellent Aβ(1−42) self-aggregation inhibition (37.5 % at 10 µM) in vitro studies [69]. The X-ray structure of TcAChE-34 complex showed that the tacrine fragment was embedded in the catalytic triad of the gorge and the quinone fragment interacts with several aromatic residues (Trp233, Phe288, Phe330, Phe331 and Trp84). Its nitrogen was associated to the carbonyl oxygen of the catalytic residue His440 through hydrogen bonding while 6-chlorine interacted strongly with Trp 432. The compound 34 also showed very good antioxidant activity in T67 human glioma cells line, good BBB penetration in ex vivo experiments on young adult Wistar rats and negligible toxicity in neuro and cerebellar granule cells in mouse [69]. These in vitro/in vivo/ex vivo results indicated that it has potential as a multi-target drug lead for further development. A novel series of tacrine-4-dimethylaminobenzoic acids was evaluated for its role against AChE for AD treatment [70]. Among all the tested analogs the analog 35a exhibited efficient hAChE (IC50 = 142 nM) and AChE-induced Aβ(1-42) aggregation inhibition (80.6% at 50 μM). It did not show cytotoxic effect on A549 cells in nanomolar concentration range. However, the analog 35b was most active against hAChE inhibition (IC50 = 19 nM) but overall drug profile of analog 35a was impressive therefore it was considered for further evaluation. A novel series of tacrine heterodimers (36 of Figure 8) was designed by combining caffeic, ferulic, and lipoic acid pharmacophore with tacrine [47,71-73]. Among them the best tacrine-caffeic hybrid [72] 36 (Figure 8) was patented showed remarkable AChE (IC50 = 0.2 nM), BChE (IC50 = 8.2 nM), self- induced Aβ(1-40) inhibitory activity (77.2% at 10 µM) and good anti-oxidant property in 2,2-diphenyl-1-picrylhydrazyl (DPPH, EC50 = 26 µM) assay. Recently, 7-MEOTA-ferulic hybrid (37 of Figure 8) bearing octylethylene linker and 2'-naphthyl fragment have been reported as multi-potent agent. It showed 19 times higher inhibition hAChE (IC50 = 22.2 nM) than tacrine (IC50 = 424 nM). It also possessed strong antioxidant property (4.29 trolox equiv.) in ORAC assay, good Aβ aggregation inhibition (65.6% at 1:1 ratio), in vitrohepatotoxicity (59.4% cell viability at 1000 µM) in HepG2 cells and good neuroprotective effect on SH-SY5Y cells at 1 µM [74]. The acridone-1,2,3-triazole hybrid (38, Figure 8) was reported for its satisfactory AChE inhibition (IC50 = 7300 nM) which was 1.5 fold stronger than rivastigmine (IC50 = 11070 nM) [75]. The methoxy group at 4-position conferred 8 fold higher activity compared to its unsubstituted analogs. This may be due to hydrogen bonding between methoxy and hydroxyl group of amino acid residue of the catalytic triad. The tacrine-trolox heterodimers [76,77] (39a-b of Figure 9) was designed by combining structural features of AChE inhibitor tacrine and antioxidant trolox to create a multifunctional agent for AD treatment. The hybrid 39a exhibited excellent AChE inhibition (IC50= 80 nM), while the hybrid 39b also showed strong hAChE inhibition (IC50 = 23.5 nM). Both hybrids possessed low in vivo hepatotoxicity and high BBB permeation in CNS. The tacrine-piperazine analog 40 (Figure 9) [78] significantly inhibited AChE (IC50 = 40 nM) which was 11 fold more active than tacrine (IC50 = 458 nM) and effectively blocked L-type calcium channels (83.93% at 50 µM). Continuing on tacrine heterodimers, Chenet al., developed tacrine-1,4-dihydropyridineheterodimers [79], among them there presentative analog 41 (Figure 10) showed remarkable rAChE inhibition (IC50 = 17 nM) which was approximately 21 fold higher active than tacrine (IC50 = 370 nM). In addition, these compounds showed neuroprotective profile and an effective L-type calcium channels blocking (83.54% at 1 µM) property [79]. Marco-Contelles et al., patented novel series of a tacrine-1,8-naphthyridine hybrids which was designed combining structural features of well-known calcium channel antagonists, nitrendipine and tacrine [80]. These hybrids were potent and selective AChE, Aβ-aggregation inhibitors, anti-oxidant, calcium ions blockers and effective neuroprotective [81-83]. Among all the tested compounds, the most selective and effective analog 42 (Figure 10) possessed excellent hAChE inhibition (IC50 = 780 nM) that was nearly 8 fold weaker than tacrine (IC50 = 100 nM) and 15 times selective over BChE (IC50 = 12000 nM). Notably, it showed high cytoprotective effect (41.5% at 1 µM) against mixture of rotenone (30 µM) and oligomycin-A (10 µM) and 58.6% at 1µM against okadaic acid (30 nM) [80]. Among the tacrine-1,8-naphthyridine hybrids, the molecules 43, 44 and 45 (Figure 10) were promising [84]. The analog 43, also known as SCR-1693 was found to be reversible, selective and noncompetitive AChEI (IC50 = 768 nM). It also showed nonselective calcium channel blocking activity (63.6% at 50 mM) [84]. Further studies on the analog 43 showed that it reduced total tau phosphorylation levels significantly in HEK293/tau cells at 5 μM, with no influence on cell viability and its efficacy was comparable with anti-alzheimer’s drug donepezil and calcium channel blocker nilvadipine at 10 μM in long term treatment [85]. It also inhibited the generation and release of Aβ in cells. Zhang et al., reported its concentration dependent selective, reversible and noncompetitive inhibitor of AChE, (IC50 = 680 nM) but not BChE in vitro and in vivo [86]. It inhibited all four types of calcium channels at 10 μM concentration non-selectively and reduced Aβ25–35-induced SH-SY5Y cell death with higher efficacy than donepezil. It was also better than donepezil and memantine in improving the Aβ25–35-induced long-term and short-term learning memory impairment in animal model. It provided protection against glutamate excite toxicity, neuronal damage and Aβ neurotoxicity which indicated that it is a potential lead molecule for AD treatment [86]. The analog 44 incorporating N,N-dimethylene side chain and fluorination at cyclohexane ring of tacrine moiety in analog 43 also showed still higher AChE inhibitory activity (IC50 = 45nM) than 43 with moderate calcium ions channel blocking property (57.2% at 50,000 nM) [84]. The other analog 45 resulted by cyclohexane ring opening of tacrine moiety was also a potent AChE inhibitor (IC50 = 270 nM) and Ca2+ ion channel blocker (49.2 % at 50 µM) [87]. The analog 46 (Figure 10) resulted by the replacement of 2-methyl and 3-carboxyl acid methyl ester by pyrazole ring of analog 42 [82]. It showed mixed-type eeAChE inhibition (IC50 =69 nM, Ki = 155 nM) with 91 times selectivity over as BChE (IC50 = 6300 nM). Additionally, it was neuroprotective (45% at 1 µM) against rotenone/oligomycin A-induced neuronal deathin SH-SY5Y neuroblastoma cells. However, it did not improve cell viability of SH-SY5Y cells damaged with Oligomycin-A (OA). The other modification at 2,3 positions of 1,8-naphthyridines by tetrahydro pyrrole and nitrile groups respectively led to analog 47 (Figure 10) [88]. It was found to be a non-competitive type eeAChE (IC50 = 25 nM, Ki = 65 nM) and hAChE (IC50 = 650 nM) inhibitor with 126 fold selectivity over hBChE (IC50 = 82000 nM). It also inhibited hAChE-induced Aβ40-aggregation (30.6% at 100 µM) effectively and monoamine oxidase-A/B in rat brain (rMAO-A; IC50 = 618 µM, rMAO-B; IC50 = 357 µM) non-selectively. Docking studies indicated that ChE inhibition of 47 was caused by 1,6-naphthyridine moiety and high MAO-inhibitory activity may be due to propargyl group. Interestingly, the compound 47 preferably bonded with PAS of the AChE enzyme which might cause inhibition of Aβ-aggregation [88].
 Figure 8: Structures of various quinoline analogs AChE inhibitors.
Figure 8: Structures of various quinoline analogs AChE inhibitors.
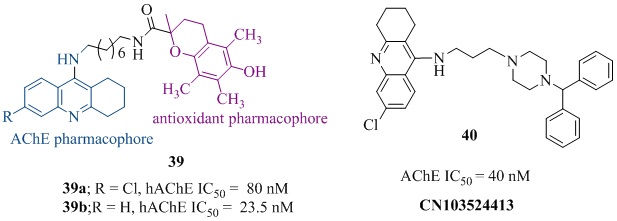 Figure 9: Structures of various quinoline analogs AChE inhibitors.
Figure 9: Structures of various quinoline analogs AChE inhibitors.
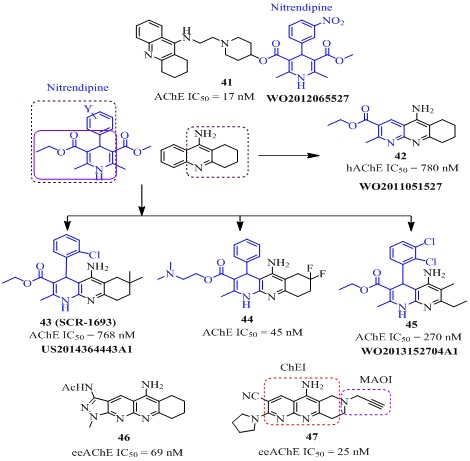 Figure 10: Acridine-1,4-Dihydropyridine/-Naphthyridine analogs AChE inhibitors.
Figure 10: Acridine-1,4-Dihydropyridine/-Naphthyridine analogs AChE inhibitors.
QUINOLINE ANALOGS
Earlier studies revealed that the FDA approved drug rivastigmine is a reversible inhibitor for both AChE and BChE. It is being used for the treatment of mild to moderate AD. It helps in cognition improvement and activities in daily life in AD patients [89]. In order to identify the active substructure of tacrine, the six member saturated ring of tacrine was replaced by more flexible open ended chains which resulted into quinoline. Therefore, quinoline and carbamate substructures were rationally integrated by molecular hybridization to produce a large number of rivastigmine-quinoline analogs, which were investigated for their AChE inhibitory activity and are discussed below in details.
Zheng et al., reported a series of novel multi-target agents by incorporating pharmacophores of three FDA-approved drugs, namely carbamyl from rivastigmine, piperidine from donepezil and propargyl amino moiety from MAO inhibitor rasagiline [90-92]. Among them the representative analog, 5-[4-propargylpiperazin-1-yl-methyl]-8-hydroxy-quinoline-dimethyl carbamate also known as HLA-20A (48 of Figure 11, Table 2) exhibited selective pseudo-irreversible AChE inhibition (IC50 = 500 nM, BChE IC50 = 42580 nM, SI = 85) [90-92]. It also showed AChE induced Aβ-aggregation inhibition, poor inhibition against iron-induced lipid peroxidation at 1–50 µM, no toxicity in human SH-SY5Y neuroblastoma cells at 10 μM in MTT assay and very weak MAO-A/B inhibition in rat brain homogenate (5% and 9% respectively at 10 μM) in vitro [93]. The rationally combining key pharmacophores of AChE inhibitors tacrine and rivastigmine with MAO-B inhibitor rasagiline, resulted into a multi-target agent 49 which is also known as M30D (Figure 11) [94]. It showed similar AChE inhibition (IC50 = 520 nM, Ki = 9.6 μM) as HLA20A (48) with 86 fold selectivity over BChE (IC50 = 44900 nM) in rat brain. The M30D was selective MAO-A inhibitor (IC50 = 0.0077 μM) and less active towards MAO-B (IC50 = 7.9 μM). The carbamyl moiety of these two analogs interacted with the active catalytic triad, whereas piperizine moiety of HLA20A interacted with the PAS and middle part of the AChE gorge and propargyl amine moiety of M30D interacted with MAO. Most importantly, carbamyl moiety in both analogs HLA20A and M30D mask the 8-hydroxylquinol oxygen which is the key atom for metal chelation in vitro studies. These both analogs get hydrolysed by AChE to release the corresponding 8-hydroxylquinol metabolites (50a-b of Figure 11). These are active metal chelators in the order Fe3+ > Cu2+ > Zn2+in brain [92]. Later Marco-Contelles et al., reported a racemic mixture of hybrid 51 (Figure 11) which was rationally designed incorporation of substructures from donepezil, propargylamine and 8-hydroxyquinoline (51 of Figure 11, Table 2) [95]. It was non selective very good eeAChE inhibitor (IC50 = 1800 nM; BChE (IC50= 1600 nM) and irreversible MAO-A (IC50 = 6.2 µM) and MAO-B (IC50 = 10.2 µM) inhibitor. It was less toxic than donepezil, exhibited BBB penetration in CNS and significantly reduced the scopolamine-induced learning deficits in adult mice in passive avoidance test. Among the reported quinoline carboxylic acid analogs [96] the compound 52, (Figure 11) was good AChE inhibitor (IC50 = 2.3 ?M) and was 3 times weaker than tacrine (IC50= 0.71 ?M). It was observed that nitrogen-containing heterocyclic rigid quinoline nucleus at the 2-position reduced the AChE inhibitory activity while more flexible nitrogen better suited with the enzyme stereochemistry. Additionally, halogen substituent on the phenyl ring improved the AChE inhibitory activity in the order F < Cl. The 6,8-disubstituted quinoline analogs [97] (53 of Figure 11)showed promising AChE inhibitory activity below 10,000 nM (IC50= 10-10,000 nM) comparable with the marketed drug rivastigmine (IC50 = 8260 nM) in recombinant human acetyl cholinesterase (rhAChE). It also showed anti-Aβ-aggregation activity and metal ion chelation property.
 Figure 11: Structures of quinoline-carbamates AChE inhibitors.
Figure 11: Structures of quinoline-carbamates AChE inhibitors.
|
Compound No |
Screening model |
Activity IC50 nM |
|
48 |
AChE |
500 |
|
49 |
AChE |
500 |
|
51 |
eeAChE |
1800 |
|
52 |
AChE |
2300 |
|
53 |
rhAChE |
100 ≈ 1000 0.001 |
Table 2: The screening model and activity IC50 of quinoline analogs.
RIVASTIGMINE ANALOGS
Earlier studies revealed that the FDA approved drug rivastigmine is a reversible inhibitor for both AChE and BuChE, is being used for the treatment of mild to moderate AD. It helps in cognition improvement and activities in daily life in AD patients. The carbamate fragment of rivastigmine binds to the catalytic triad of AChE enzyme for much longer than the acetate fragment of ACh during the hydrolysis of ACh and is responsible for its AChE activity for longer duration. Additionally, rivastigmine shows relatively low protein-binding affinity and does not interfere in other medication for concurrent illness. The enzyme AChE exists in several forms and rivastigmine exhibits selectivity for the G1 form which progressively increases with advances of AD, than G4 form which is found in abundance in normal human. Hence, it plays more important role in hydrolyzing ACh at cholinergic synapses during the progression of AD [89]. All these unique qualities of rivastigmine increases its bio-efficacy and attracted researcher’s attention to explore its derivatives. Some potent rivastigmine or carbamate analogs appeared in last five years (2011-2015) as anti-Alzheimer’s agents, are discussed below.
Various substituted 1,2,3,4-tetrahydroquinolin-6(or-7)-yl carbamates (54-57 of Figure 12, Table 3) have been reported as anti-alzheimer’s agents [98-101]. Their design and synthesis was based on a systematic Virtual Screening (VS) experiments, consisting of the development of 3D-pharmacophore models, screening of virtual library, synthesis and pharmacology. The initial lead compound 2-chlorophenylcarbamic acid 1-benzyl-1,2,3,4-tetrahydroquinolin-6-yl (54, Figure 12) showed IC50 value 3.31 ?M [100]. Considering the high aromatic content at the active site gorge and the dominating hydrophobic interactions of AChE with its inhibitors, the modifications were targeted at site-1 and site-2of the lead compound 54. The comparatively more hydrophobic propargyl and 2,4-dichlorobenzyl groups were the two highly suited groups at site-1 as R1. The attempted major modifications at the site-2 of the lead compound 54 where both substituted aromatic and long-chain (C6–C8) alkyl groups were incorporated which led to the invention of a series of novel compounds 55 and 56 (Figure 12). Among these molecules the three promising compounds, 55 (AChE, IC50 = 0.70?M) showed almost comparable AChE inhibitory activity in Swiss albino mice brain (mAChE) with the drug rivastigmine (mAChE, IC50 = 1.11 ?M) in vitro studies [100]. Interestingly, through docking studies in the substituted 1,2,3,4-tetrahydroquinolin-7-yl carbamates analogs 56 (Figure 12) [101] were identified to be more potent than their analogs 55. Among these the molecule 56 (mAChE, IC50 = 0.10 ?M) possessing benzyl group on the carbamate end exhibited 11-fold better AChE inhibitory activity compared to the currently used drug rivastigmine and almost equal or even slightly better AChE inhibitory activity compared to the drug tacrine. The compound 56 has also shown potential towards the improvement in learning in mice in behavioral study which was almost comparable to the drug rivastigmine and tacrine in vivo studies. The molecules 55-57 were evaluated for in vivo passive avoidance test and aldicarb-sensitivity assay, led to the discovery of orally active novel AChEIs which improved scopolamine-induced cognition impairment in Swiss male mice. The % learning improvement of molecules 56 was comparable to donepezil but weaker than tacrine (Figure 12). The 1,2,3,4-tetrahydroisoquinolin-6-yl carbamate analog 58 (Figure 12) showed potent AChE inhibition (IC50 = 100 nM) which was approximately 10 times higher active than rivastigmine (IC50 = 1030 nM) in rat brain and efficiently regulated L- type calcium channel inhibitory activity (53% at 50 µM) [102]. The N-substituted carbamoyl derivative 59 (Figure 12) showed very good AChE inhibition (IC50 = 21800 nM) which was nearly 22 fold higher than rivastigmine (IC50 = 501000 nM) [103]. This compound was highly lipophilic with lipophilicity values (log k = 1.09, log P = 9.13). The 4-methyl-2-oxo-2H-chromen-7-yl phenyl carbamate (60 of Figure 13) exhibited strong mAChE inhibition (IC50 = 13.5 nM) which was 20 times more selective over BChE and also evaluated for memory testing in scopolamine-induced amnesia [104]. The incorporation of AChE inhibitory moiety-carbamate with anti-Aβ aggregation/metal chelating pharmacophores thioflavin and deferiprone moieties resulted in compounds 61 and 62 respectively (Figure 13, Table 3). The thioflavin-carbamate 61 was an irreversible AChE inhibitor (IC50 = 20900 nM) while the deferiprone-carbamate 62 was reversible and mixed non-competitive/competitive type AChE inhibitor (IC50 = 48800 nM). These two prodrugs were nontoxic below 200 µM and showed very good biological compatibility data for drug-likeness. A novel phenylcarbamate derivative meserine 62 (Figure 13), showed significant in vitro hAChE inhibitory activity (IC50 = 274 nM) and significantly improved spatial learning and memory deficits in scopolamine-induced dementia in adult mice at 1 mg/kg, i.p. dose. Furthermore, meserine reduced APP level by 28% at 7.5 mg/kg dose in 3 weeks and Aβ42 level by 42% in APP/PS1 transgenic mouse [105]. Recently Bohn et al., reported selective central AChE inhibitors among them the bio-oxidizable prodrug 63 (Figure 13) was almost inactive against AChE (IC50 > 1mM) in peripheral system [106]. However after crossing the BBB (Blood Brain Barrier), the prodrug 63 oxidised in CNS to produce prodrug 64 which further activated a central cholinergic system by carbamylation of AChE with remarkably high activity (IC50 = 20 nM) and released a metabolite 5-hydroxyl quinolinium which transported out of the brain easily. Overall, in vivoand ex vivo studies indicated that the “bio-oxidizable” prodrug approach could be a promising strategy for rational designing of selective central AChE inhibitors.
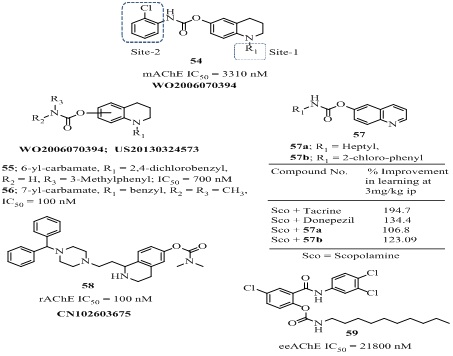 Figure 12: Structures of rivastigmine-carbamates analogs.
Figure 12: Structures of rivastigmine-carbamates analogs.
|
Compound No. |
Screening model |
Activity IC50 nM |
|
54 |
mAChE |
3310 |
|
58 |
rAChE |
100 |
|
59 |
eeAChE |
21800 |
|
60 |
mAChE |
13.5 |
|
61 |
eeAChE |
20900 |
|
62 |
eeAChE |
48800 |
|
Meserine- 62 |
hAChE |
274 |
|
63 |
hAChE |
1000000 |
|
64 |
hAChE |
2 |
Table 3: The screening model and activity IC50 of rivastigmine analogs.
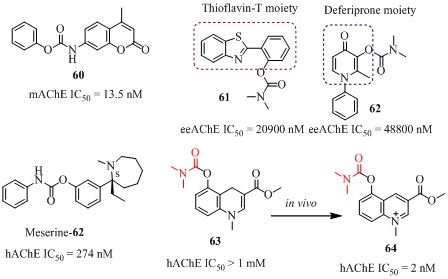 Figure 13: Structures of carbamate analogs.
Figure 13: Structures of carbamate analogs.
DONEPEZIL ANALOGS
The FDA approved anti-Alzheimer’s drug, donepezil (Aricept) is cis1-benzil-4-[5,6-dimethoxy-(1-indanone)-2-yl]-methylpiperidine hydrochloride (Figure 14, Table 4), which is a versible, non-competitive and very effective AChE inhibitor (IC50 = 5.7 nM) with less toxicity [107,108]. This drug is clinically used for the treatment of AD in patients with mild, moderate and severe stages of cognitive impairment at maximum 23mg/day dose [107]. The phenomenon Deuterium Kinetic Isotope Effect (DKIE) has been used on donepezil drug to improve its Pharmacokinetics (PK), Pharmacodynamics (PD), and toxicity profiles [109] viz increased half-life, reduced metabolic rate and thus making it more effective and safer drug. Seeing the high potency and low toxicity of donepezil, later researchers focused on designing new multi-potent AChE inhibitors by combining the structural features of donepezil with bioactive chemical entities. Thus the donezepil was considered a molecule two structural fragments represented by site-1 and site-2 connected through a spacer. The majority of novel donezepil analogs were achieved by structural modifications in these two sub-structures and the spacer. These novel donepezil analogs are discussed below [108].
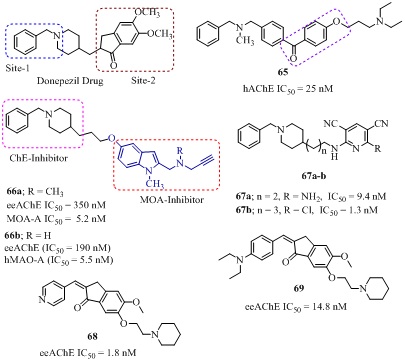 Figure 14: Structures of donepezil derived AChE inhibitors.
Figure 14: Structures of donepezil derived AChE inhibitors.
|
Compound No. |
Screening model |
Activity IC50 nM |
Compound No. |
Screening model |
Activity IC50 nM |
|
65 |
hAChE |
25 |
76 |
eeAChE |
42 |
|
66a |
eeAChE |
350 |
77 |
AChE |
52 |
|
66b |
eeAChE |
190 |
78 |
AChE |
4.1 |
|
67a |
AChE |
9.4 |
79 |
AChE |
48 |
|
67b |
AChE |
1.3 |
80 |
AChE |
8.9 |
|
68 |
eeAChE |
1.8 |
81 |
AChE |
0.44 |
|
69 |
eeAChE |
14.8 |
82 |
AChE |
27 |
|
70 |
eeAChE |
0.3 |
83 |
AChE |
55 % inhibition |
|
71 |
eeAChE |
4900 |
84 |
eeAChE |
910 |
|
72 |
eeAChE |
1200 |
85a |
hAChE |
160 |
|
73 |
eeAChE |
1600 |
85b |
hAChE |
0.0016 |
|
74 |
eeAChE |
8600 |
86 |
AChE |
0.041 |
|
75 |
eeAChE |
420 |
87 |
eeAChE |
0.01 |
Table 4: The screening model and activity IC50 of donepezil analogs.
The donepezil analog 65 having benzophenone scaffold bearing N,N-diethylaminopropoxy group (site-2) connected with benzyle fragment (site-1) of donezepil [110] showed highly selective AChE inhibition (IC50 = 25 nM, Ki = 24.3 nM, SI = 1372) comparable to the donepezil (IC50 = 23 nM). It also showed AChE-induced Aβ(1-40)-inhibition (34.3% at 250 µM). The molecular modeling studies indicated that benzophenone scaffold bonded with arromatic residues of the AChE gorge through π-π interaction and N,N- diethylaminopropoxy group suitably fit in the PAS pocket of the enzyme. Marco Contelles et al., reported a series of donepezil- indolylpropargyl amines derivaties, designed by combining benzylpiperidine moiety of the AChE inhibitor donepezil and indolyl propargylamino moiety of the MAO inhibitor [N-(2-propynyl)-2-(5-benzyloxy-indolyl) methylamine] connected through an oligomethylene linker of apropriate length [111]. Among these the analog 66a, also known as ASS234 (Figure 14) exhibited good AChE inhibitory activity (IC50 = 350 nM) with no selectivity over BChE (IC50 = 430 nM). It also irreversibly inhibited MAO in human (MAO-A IC50 = 5.2 nM, MAO-B = 43 nM) [111] and was almost as effective as clorgyline (Kintact/K1 = 3 ×106 min−1 M−1) [112]. The N-dealkylation of the propargylamine moietyof analog 66a resulted into analog 66b (Figure 14) which exhibited excellent eeAChE (IC50 = 190 nM) and esBChE (IC50 = 830 nM) inhibition along with strong MAO-A (IC50 = 5.5 nM) and MAO-B (IC50 = 150 nM) inhibition [113]. The molecular modeling studies indicated that the analogs 66a-b interacted at both sites CAS and PAS of the AChE. In addition, both 66a-b inhibited AChE induced Aβ-aggregation and Aβ(1-42) self-aggregation. They significantly reduced Aβ(1-42)-induced toxicity in SH-SY5Y neuroblastoma cells in human. They possessed free-radical scavenger capacity and blood-brain barrier penetration ability (Pe = 10.3 × 10−6 cm/sec) in vitro [114]. The molecular hybridization of N-benzylpiperidine moiety of donepezil and substituted pyridine moiety, resulted into pyridonepezil analogs (67a-b of Figure 14) [115,116]. Where, the 2-amino-6-((3-(1-benzylpiperidin-4-yl)propyl)amino)pyridine-3,5-dicarbonitrile (67a) exhibited remarkable hAChE inhibitory activity (IC50 = 9.4 nM) with medium level selectivity (SI = 703) over BChE (IC50 = 6600 nM) [115]. The 6-chloro-pyridonepezil (67b) was potent hAChE inhibitors (IC50= 13 nM) 625-fold more selective over hBChE (IC50 = 8130 nM) and equipotent to donepezil (IC50 =10 nM) [116]. The both analogs 67a-b showed BBB-PAMPA penetration (67a; Pe = 20.8, 67b; Pe = 6.99 cm/s) by passive diffusion. The theoretical absorption, distribution, metabolism, and excretion (ADME) analysis of both anlogs 67a-b showed significant values for the analyzed properties and drug-like characteristics based on Lipinski’s rule of five.The donepezil-indanone 68 (Figure 14, Table 4) exhibited 14-fold more potent AChE inhibition (IC50 = 1.8 nM) than donepezil and 5248 fold more selective over BChE [117]. Furthermore, 68 also exhibited significant metal-chelating (Fe+3, Cu+2, Zn+2) capacity. The donepezil-indanone analog 69 (Figure 14) showed mixed type AChE inhibition (IC50 = 14.8 nM) 2 times stronger than donepezil (IC50 = 38.2 nM) and 14 times stronger than tacrine (IC50 = 109 nM) [118]. In addition, it also exhibited self induced Aβ aggregation inhibition (85.5% at 20 µM) in Thioflavin T fluorescence assay (ThT), antioxidant activity (3.70 trolox equiv.) in ORAC-FL assay and ability to cross BBB (Pe = 1.4574 cm/s) in CNS.
A large number of donepezil-coumarin analogs have been evaluated for inhibition of AChE for the treatment of AD. Among them the analog 70 (Figure 15, Table 4) with 6-nitro-substituent was a mixed type potent AChE inhibtor (IC50 = 0.3 nM) with highest selectivity (SI = 26300) and 46-fold higher potentency than donepezil (IC50 = 14 nM) [119]. The donepezil-coumarin analog 71 (Figure 15) connected through with piperzine spacer at 4-position of coumarin also found to be moderate AChE inhibitor (IC50 = 4900 nM) [120]. The other donepezil-coumarin analog 72 (Figure 15) where benzyl piperazidine was connected with 4-position of coumarin through an acetamide linker, exhibited good eeAChE inhibitory activity (IC50 = 1200 nM) with 37 fold selectivty [121]. The molecular modeling of analog 72 indicated that π-cation interaction with benzylpiperidine moiety with Phe330 in CAS and additional π-π interaction between coumarin moiety and Trp279 in PAS of AChE, was responsible for its duel binding site ability. The other donepezil-7-hydroxycoumarin hybrid 73 (Figure 15) connected through an amidic linker, was mixed-type AChE inhibitor (IC50 = 1600 nM, Ki = 1.32 µM) with 26 fold selectivity and also significantly protected PC12 neurons against H2O2-induced cell death (P < 0.001) at 1 µM [122]. The other donepezil-3-coumarin analog 74 also known as AP2469 (Figure 15) connected through ethyl-dimethylamine linker showed significant ChE inhibtion (AChE IC50 = 8.6, BChE IC50=124000 nM respectively) [123]. Aditionally, it showed BACE-1 inhibition (IC50 = 6490 nM), anti-Aβ(1-42) self-aggregation (50% inhibition at 21.7 µM), anti-Aβ(1-42) oligomer toxicity (50% at 7.50 µM) in SH-SY5Y cells and in THP-1 cells (50% at 8.18 µM) along with intrinsic antioxidant activity (1.6 kROO. (mol/L.sec)1) [123]. The combination of structural features of analog 73 and 74 resulted into a more flexible analog 75 (Figure 15) with balanced hydrophilicity/lipophilicity ratio [124]. It possessed excellent eeAChE (IC50 = 420 nM) and selective rMAO-B (IC50 = 410 nM) inhibitory activity profile. Furthermore, it exhibited BBB-permeabilty by by passive diffusion, neuroprotective agent against H2O2-induced oxidative stress (25% at 10 µM) and no cytotoxicity in cell line SH-SY5Y up to 50 μM after 72 h [124].
 Figure 15: Structures of donepezil-caumarin analogs.
Figure 15: Structures of donepezil-caumarin analogs.
The molecular hybridization of ChE inhibitor donepezil and the antioxidant ebselen resulted into a multi-target-directed ligand 76 (Figure 16) for the treatment of AD [125]. The analog 76 exhibit potent AChE inhibitory activity (IC50 = 42 nM) in Electric eel and in human (IC50 = 97 nM). Additionally, it possessed good AChE-induced Aβ(1−40) aggregation inhibition (21.4 % at 100 µM) activity and glutathione peroxidase-like activity (ν0 = 123.5 μM/min). This analog 76 did not show acute toxicity at doses of up to 2000 mg/kg in mice and was able to penetrate the central nervous system [125]. The several combinations of benzylpyridinium salt with benzofuran/indol have been investigated for their role in AChE inhibition. Among them the conjoined 5,6-dimethoxy benzofuranone and substituted benzylpyridinium bromide resulted into analog 77 (Figure 16) which possessed strong AChE inhibitory activty (IC50 = 52 nM) with low selectivity (SI = 31) over BChE (IC50 = 1620 nM) and approximately 2 fold weaker than donepezil (IC50 = 31 nM) [126]. While the analog 78 (Figure 16) where the dimethyl-substituted benzylpyridinium bromide directly connected with benzofuran was 7 fold stronger than donepezil (IC50 = 4.1 nM) [127]. The expension of benzofuran ring and bromo-substitution in benzylpyridinium moiety of analog 77 resulted into analog 79 (Figure 16, Table 4) which inhibited AChE (IC50 = 48 nM) 2 fold weaker than donepezil [128]. The structural features combination and modification of analogs 77 and 79 resulted into benzyl pyridinium-4-isochromanone hybrid (80 of Figure 16) exhibited excellent AChE activity (IC50 = 8.9 nM) [129]. Among the series of donepezil-indolinone hybrids the analog bearing 2-chlorobenzylpyridinium moiety (81 of Figure 16, Table 4) showed most potent AChE inhibition (IC50 = 0.44 nM) which was 32 fold more active than donepezil. The molecular studies showed that indoline part bounded with PAS and N-benzylpyridinium fragment inteacted with the CAS of AChE [130].
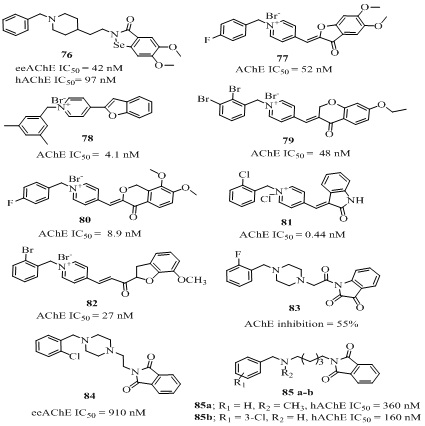 Figure 16: Structures of donepezil analogs as AChE inhibitors.
Figure 16: Structures of donepezil analogs as AChE inhibitors.
The benzylpyridinium-chalconoids hybrid 82 (Figure 16 ) bearing N-(2-bromobenzyl) moiety and 7-methoxy substituent on the benzofuran ring exhibited excellent AChE inhibitory activity (IC50 = 27 nM) [131]. The analog 83 (Figure 14) showed inhibitory activity (55% inhibition at 1266.84 ChE (U/gm) better than donepezil, mainly due to its additional 3-oxo group and 2-flourobenzyl substitution [132]. The benzyl moiety connected with isoindoline-1,3-dione through a piperazine spacer resulted into analog 84 (Figure 16) showed AChE inhibition (IC50 = 910 nM) in Electric eel weaker than donepezil (IC50 = 140 nM) [133]. The electron withdrawing groups like Cl, F and NO2 in displayed best effect at position ortho and para of the phenyl ring. Continuing the series Guzior et al., reported isoindoline-1,3-dione analogs 85a-b (Figure 16), where unsubstitution 5 atom spacer favored the potency of the molecule against AChE (85a-b; hAChE IC50 = 360,160 nM respectively) noncompetitive mode and were able to cross the BBB to reach its biological target in CNS [134,135].
In view of the biological and medicinal properties of oxoisoindoline analogs, the donepezil-oxoisoindoline hybrid was evaluated against AChE inhibition (IC50 =41040 nM) [136]. The donepezil-hydrazinonicotinamide analog (87 of Figure 17, Table 4) designed by combining structural features of donepezil and hydrazine nicotinate moiety, showed AChE inhibitory activity (IC50 = 10870 nM) with 249 selectivity [137].
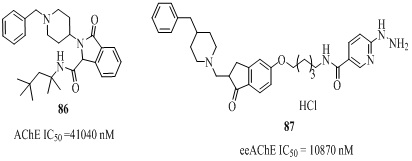 Figure 17: Structures of donepezil derived AChE inhibitors.
Figure 17: Structures of donepezil derived AChE inhibitors.
HUPERZINE ANALOGS
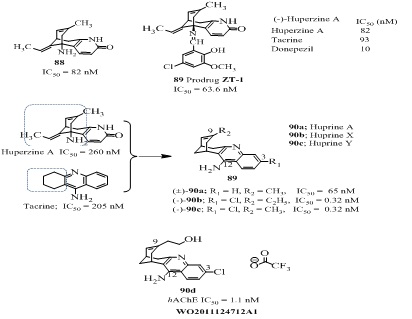 Figure 18: Huperzine analogs with their AChE IC50 values.
Figure 18: Huperzine analogs with their AChE IC50 values.|
Compound No. |
Screening model |
Activity IC50 nM |
Compound No. |
Screening model |
Activity IC50 nM |
|
88 |
AChE |
82 |
94 |
AChE |
17.5 nM |
|
89 |
AChE |
63.6 |
95 |
AChE |
6.7 nM |
|
90 a |
AChE |
65 |
96 |
AChE |
4.2 nM |
|
90 b |
AChE |
0.32 |
97 |
AChE |
3.1 |
|
90 c |
AChE |
0.32 |
98 a |
hAChE |
0.42 |
|
90 d |
AChE |
1.1 |
98 b |
hAChE |
0.31 |
|
91 |
AChE |
0.61 |
99 |
hAChE |
2.61 |
|
93 |
AChE |
1.07 |
100 |
rAChE |
9.0 |
Table 5: The screening model and activity IC50 of huperzine analogs and huprine heterodimers.
The molecular hybridization of two known active molecules namely tacrine and huperzine A, resulted into a new lead molecule huprine (90; Figure 18) with increased potency and extended sites for functional modification.
Among more than 40 synthesized analogs, the racemic huprine A (90a; IC50 = 65 nM), enantiopure, (–)-huprine X (90b IC50 = 0.32 nM) and Y (90c) (IC50 = 0.32 nM; Figure 18, Table 5) were promising. The analogs 90b and 90c were 800- and 640-fold more potent than their parental molecules (–)-huperzine A and tacrine, (IC50 = 260 nM; 205 nM respectively) [144]. The high-affinity of huprine X towards inhibition of AChE and low effective dose of rac-huprine Y (90c) in ex vivo studies (ID50 = 1.09 µmol/kg) indicated that high activity resulted in a much lower dose, may avoid the toxicity problems associated with higher dose of tacrine. The huprine A, X and Y showed cognitive-enhancing properties and their effects on the regulation of several neurochemical processes related to AD in triple transgenic mice (3xTg-AD) [145]. Therefore these were selected for further lead optimization. A series of Huprine X with diverse modifications at position-9 and position-12 was evaluated for AChE inhibitory activity [146-148]. Among them the representative analogs (90d of Figure 18) inhibited AChE at nano molar (hAChE IC50 = 1.1 nM) with high selectivity towards AChE (SI = 1130) in human and low dissociation constant (KD = 0.87 nM). The molecular studies suggested that the terminal group of alcohol in molecule 90d forms additional favorable hydrogen bonds with the catalytic Ser203 and Gly122 of the oxy anion hole. Its LD50 in Swiss albinomice was 40 mg/kgip and it was able to cross the BBB at doses above 15 mg/kg [147].
The steric bulky substituent’s at position-9 decreased AChE inhibitory potency, as bulk substituent it did not fit in the enzyme’s binding pocket. It was proposed that aniline group interacted with residues Thr83, Trp86, Tyr133, Ser125 and Glu202 in catalytic gorge of the enzyme through hydrogen bonding with the help of key water molecules. Therefore the functionality at position-12 decreased the potency [148]. Hence the substitution at position 9 provides the opportunity to generate a linker that will not disrupt the water network and that can reach the peripheral site without making steric clashes with gorge residues. This led to the development of dual binding huprine-heterodimer inhibitors by coupling huprine with heterocyclic ligands, which are discussed below.
Huprine-Heterodimers
 Figure 19: Huprine-heterodimers with their AChE inhibitory activity.
Figure 19: Huprine-heterodimers with their AChE inhibitory activity.The huprine-coumarin heterodimers [146,149] (92; Figure 19, Table 5) was another potent AChE inhibitor with dissociation constant at pico molar level. It strongly inhibited Aβ peptide aggregation at 330 pM to 67 nM concentrations. The dual binding site huprine-heterodimers were also developed by functional modification at amino group at position-12. Huprine in hybrids were designed by joining huprine with sub structure of already known Aβ and tau anti-aggregation agent hydroxyl anthrax quinone. Among these hybrids the rac-93, (Figure 19, Table 5) exhibited potent AChE inhibitory activity (IC50 = 1.07 nM) along with BACE-1, dual Aβ-42 and tau anti-aggregation properties with efficient brain permeability [150]. The homodimers of (+)-(7R,11R)-huprine Y, connected through hexa-, octa-, deca-, dodeca-methylene or p-phenylene-bis (methylene)-spacer have been reported [151]. Among them the most potent AChE inhibitor 94 (AChE IC50= 17.5 nM) was 29 times weaker than its parent molecule (+)-(7R,11R)-huprine Y (AChE IC50 = 0.6 nM). These bis(+)-huprines also acted as potent trypanocidal agents. The analog 94 with dodecyl spacer was less cytotoxic towards mammalian cells and inhibited Caenorhabditis elegans (C.elegans) growth over rat L6 cells more selectively than the huprine Y. All the bis(+)-huprines Y showed ability to cross the BBB [151].
Huprine-shogaol hybrid (95 of Figure 20) where huprine Y was connected with a substructure of already known antioxidant through different spacers were explored for their AChE inhibitory activities [152]. The resulting analog viz racemic shogaol–huprine hybrid 95 (IC50 = 6.7 nM) was nearly 10 times less potent than the racemic huprine Y (IC50= 0.7 nM). The presence of an additional basic nitrogen atom at the phenyl ring did not influence the hAChE inhibitory activity significantly. Additionally, these hybrids also acted as potent antioxidant agent, Aβ-42 and tau anti-aggregating agents. The huprine-levetiracetam hybrids (Figure 20) were derived by medicinal chemical hybridization of either (7S,11S)- or (7R,11R)-huprine Y and substructure of antiepileptic drug (S)-levetiracetam, through a heptamethylene spacer [153]. Among them the hybrid (2S,7S,11S)-96 (IC50= 4.2 nM) was six times less potent AChE inhibitor than its parental molecule 7S,11S-Hup Y (IC50 = 0.74 nM). The replacement of huprine Y by acridine led to the acridine-levetiracetam hybrids [154] where the racemic hybrid 97 (IC50 = 3.1 nM, Figure 20) found to be 2 times more potent AChE inhibitor than its parent structural part 6-Chloro-THA (IC50 = 5.9 nM). This analog was equipotent with huprine-levetiracetam hybrid 96 (IC50= 4.2 nM). Additionally, these hybrids 96 and (±)-97 moderately inhibited Aβ-42, tau aggregation and neuro-inflammation along with ability to cross blood barrier.
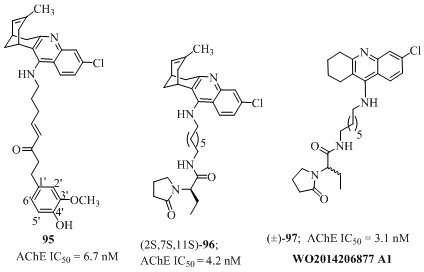 Figure 20: Huprine-shogaol, -levetiracetam analogs as AChE inhibitors.
Figure 20: Huprine-shogaol, -levetiracetam analogs as AChE inhibitors.The huprine-acridine heterodimers 98a-b (Figure 21) were designed by combining the two active AChE inhibitors namely racemic huprine Y and tacrine or 6-chlorotacrine through 6−10 methylene units or a 4-methyl-4-azaheptamethylene spacer [155,156]. These analogs were claimed to interact simultaneously with both catalytic and peripheral sites of AChE (98a-b; Figure 21). Among these potent AChE inhibitors, the molecule (±)-98a and (±)-98b (IC50 = 0.42, 0.31 nM respectively) were 1.6 and 2 times more potent respectively than the parent structure part (±)-huprine Y (IC50 = 0.69 nM). Furthermore, these molecules also showed Aβ anti-aggregation, β-secretase and BACE-1 inhibition along with blood-brain barrier permeability shown in vitro and ex vivo experiments with OF1 mice. The molecular hybridization of two potent AChE inhibitors namely donepezil and huprine Y, resulted in a series of analogs, among them the enantiomeric pure (-)-(7S,11S)-99 also known as AVCRI104P4 (IC50 = 2.61 nM, Figure 21) was 8 times more potent than donepezil (IC50 = 21.4 nM) but 6 times weaker than the huprine Y (IC50 = 0.43 nM ) with high selectivity towards hAChE over hBChE (SI = 134) [157]. Moreover, in vivo efficacy studies of this hybrid showed that strong AChE inhibition but very less effect on Aβ aggregation (29% at 10 µM) either in APPSL mice or in C. elegans and BACE-1 (IC50 = 11000 nM). The improved short-term memory was observed in transgenic APPSL mice. The heterodimer of dimethoxyindanone and hupyridone (RS,S)-100 (Figure 21) with tetramethylene linker exhibited high AChE inhibitory activity (IC50 = 9.0 nM) in rats. It was equally potent to donepezil (IC50 = 5.7 nM) and 7 fold more active than Huprine A with 100,000 selective over BChE (IC50 = 1 mM) [158].
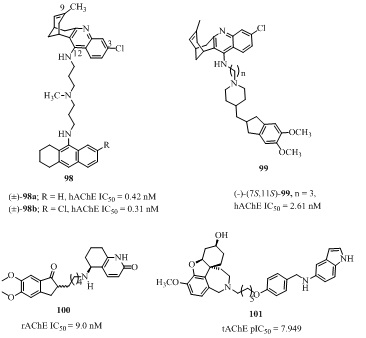 Figure 21: AChE inhibitors with their AChE IC50 values.
Figure 21: AChE inhibitors with their AChE IC50 values.GALANTHAMINE ANALOGS
Galantamine (Figure 2) is an alkaloid isolated from the bulbs and flowers of Galanthus woronowii in plant family Amaryll idaceae [159] and later synthesized by several researchers [160,161]. It is a reversible competitive and selective AChE inhibitor. It also inhibited human nicotinic ACh receptors (nAChRs) in AD patients, but has not been explored well due to its toxicity factor [162,163]. In 2015, Doytchinova et al., reported the galanthamine-indole heterodimer 101 (Figure 21, Table 6) as a potent AChE inhibitor against enzyme rhAChE experimentally (IC50 = 11 nM) [164] which is 82 times stronger than galanthamine (IC50= 1070 nM) against AD. The galanthamine moiety binds to the CAS and the indole moiety binds to PAS of AChE enzyme. Additionally, it was able to block the Aβ deposition on AChE.
|
Compound No. |
Screening model |
Activity IC50 nM |
Compound No. |
Screening model |
Activity IC50 nM |
|
101 |
tAChE |
7.949 |
108 |
eeAChE |
360400 |
|
102 |
AChE |
7300 |
109 |
rAChE |
90 |
|
103 |
AChE |
210 |
110 |
eeAChE |
40 |
|
104 |
eeAChE |
64000 |
111 |
AChE |
2160 |
|
105 |
eeAChE |
1100 |
112 |
hAChE |
20 |
|
106 |
eeAChE |
157300 |
113 |
eeAChE |
1500 |
|
107 |
hAchE |
130 |
Table 6: The screening model and activity IC50 of galanthamine analogs.
NOVEL SMALL MOLECULES
Apart from the above mentioned homo and hetero dimers, many other small molecules have been explored for the treatment of AD. Some recently reported potent small AChE inhibitors are discussed here.
A series of naturally occurring phenolic acids (derivatives of caffeic acid, rosmarinic acid and trolox) are known for their anti-oxidant properties have been conjugated with choline to develop AChE inhibitors with good antioxidant properties. The resulting hybrid 102 (Figure 22) possessed very good AChE inhibition (IC50 = 7300 nM) along with anti-oxidant activity (EC50 = 303 µM) [165]. Among the various reported chalcone derivative [166,167] the 103 (Figure 22) was a mixed-type inhibition against AChE (IC50 = 210 nM) which was 50-fold more potent than the drug rivastigmine (IC50 = 10540 nM) [168]. The 1-2-(benzo[d]thiazol-2-yl)-3-heteroarylacrylonitriles 104 (Figure 22, Table 6) was found to be a competitive AChE inhibitor (IC50 = 64000 nM) in Electric eel [169]. The isoindoline-1,3-dione derivative 105 (Figure 22) with 7 methylene linker moderately inhibited eeAChE (IC50 = 1100 nM) and weakly inhibited Aβ plaque formation (39.4% at 80 µM) [170]. The pyrazole derived small molecules, 1-[3-Methyl-5-(2,6,6-trimethyl-cyclohex-1-enyl)-4,5-dihydro-pyrazol-1-yl]-ethanone (106 of Figure 22) also showed average inhibitory activity against AChE (IC50 = 157300 nM) and selective inhibitory activity towards BChE (IC50 = 46400 nM) enzyme. A series of 1H-benzimidazole derivatives was evaluated against AChE inhibition [171]. Among them the molecule 107 (Figure 22) was most active (IC50 = 130 nM). The lipophilic group (Cl) substitution on 5-position of benzimidazole scaffold may contributed in higher activity. The resveratrol derivative 108 (Figure 22) exhibited significant AChE inhibition (IC50 = 36040 nM) [172]. Additionally, it also showed anti-oxidant activity using fluoresce in (ORAC-FL, 4.72 trolox equiv./μmol), self-induced Aβ aggregation (IC50 = 7.56 µM) and MAO-A/B inhibition (IC50 = 8.19, 12.16 µM respectively). It acted as potential antioxidants and biometal chelators such as Cu+2 and Fe+2. It was able to penetrate the BBB in vitro and did not show any toxicity at up to doses 2000 mg/kg in mice [173]. Thus indicating it’s potential as lead molecule for detailed investigations. The aloe-emodin derivative 109 (Figure 22) containing quaternary pyridinium fragment found to be non-competitive or mixed type of AChE inhibitor (IC50 = 90 nM) on rat cortex homogenate which was better than tacrine (IC50 = 260 nM) [173]. The novel α,β-unsaturated carbonyl compound 110 (Figure 22) has been evaluated for its AChE inhibition (IC50 = 40 nM) and free radical scavenger activity (DPPH IC50 = 20.40 µM) [174] where the diethoxymethyl group at position 4 and presence of piperidone moiety might have contributed in its strong AChE inhibition. The seleno-dihydropyrimidinone 111 (Figure 22) exhibited very good AChE inhibtroy activity (IC50 = 2160 nM) and Fe+2 chelating activity (EC50 =23.79 μM) [175]. The reported compound showed high antioxidant activity 521.81 mg of Ascorbic Acid Equivalents (AAE) per gram of the compound at 100 μM. It was also showed favorable pharmacokinetics profile, including solubility and permeability. The (Z)-acrylonitrile analog 112 (Figure 22, Table 6) bearing trimethoxy group showed efficient AChE inhibition, with IC50 values of 0.20 μM in human [176] where the methoxy group at the 4-position of phenyl ring has been considered essential for AChE inhibition. The 3,4-dihydroxybenzoic acid derivative 113 (Figure 22) found to be highly selective non-competitive or mixed type of eeAChE inhibitor with low 1.5 μM Ki values and showed very good metal (Fe+2, Cu+2, Zn+2) chelating properties [177]. The reported molecule also exhibited good antioxidant activity as remarkable radical scavenging ability (EC50= 0.12 μM) which was higher than trolox (EC50 = 0.15 μM) in 2,2-Diphenyl-2-Picrylhydrazyl (DPPH) assay.
 Figure 22: Small molecule AChE inhibitors.
Figure 22: Small molecule AChE inhibitors.
NOVEL HETEROCYCLIC MOLECULES
A number of heterocyclic molecules incorporating various heterocyclic rings, such as imidazole, pyrazole, thiazole, thiophene, pyridine, pyrimidine and triazine have been evaluated against AChE inhibition in search of anti-Alzheimer’s agents. Among them some potent heterocyclic molecules reported in last 5 years are described here.
The tetra cyclic thienopyrimidine 114 (Figure 23, Table 7) was a mixed-type inhibition of AChE (IC50 = 5.69 μM) in human [178]. The heterocyclic scaffolds benzotriazinone 115 (Figure 23) linked to basic amine through a linear alkyl chain, emerged as a potent AChE inhibitor (IC50 = 1.5 μM) in Electric eel and Aβ-anti-aggregation agent (IC50 = 1.4 μM) [179]. Different thiazolo-pyrimidine analogs have been reported as AChE inhibitors [180-182] where the analog 116 was most active with 89.8% AChE inhibition at 10 ?M concentration [181]. The 3-amino alkane acylamino-rutaecarpine derivative 117 (Figure 23) also showed strong AChE inhibition activity (IC50 = 0.6 nM) in Electric eel and high selectivity 3092 over BChE along with efficient self/AChE induced Aβ-aggregation inhibition, anti-oxidant and metal chelation activity [183]. The 2-(2-indolyl-)-4(3H)-quinazolines derivate 118 (Figure 23) was ring open analog of rutaecarpine alkaloid [184]. The pharmacological evaluation of molecule 118 revealed that it was a strong and selective AChE inhibitor (IC50 = 20.97 nM, SI = 349) in Electric eel. The structurally modified heterocyclic molecule 119 (Figure 23) was also good AChE inhibitor (IC50 = 142 nM) in human enzyme which was approximately 2 fold less active than tacrine drug (IC50 = 71.5 nM) [185]. The 2,6-disubstituted pyridazinone [186] 120 (Figure 23) proved highly potential AChE inhibitor with very low IC50 value 28 nM in homogenate of rat cerebral cortex. This lead molecule should be considered for further pharmacological development. Among the various known thiadiazol derivatives [187-190] the (1,3,4-thiadiazol-2-yl) benzene-1,3-diol derivative 121 ( Figure 23) was a mixed type and potent AChE inhibitor (IC50 = 53 nM) with 950 fold selectivity index over BChE (IC50 = 50200 nM) in Electric eel [188].
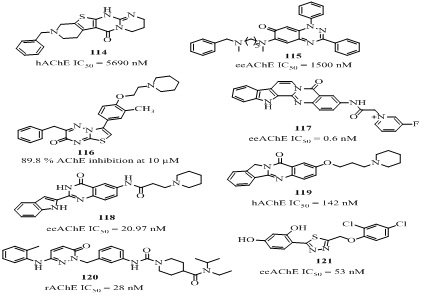 Figure 23: Heterocyclic AChE inhibitors.
Figure 23: Heterocyclic AChE inhibitors.
|
Compound No. |
Screening model |
Activity IC50 nM |
Compound No. |
Screening model |
Activity IC50 nM |
|
114 |
hAChE |
5690 |
127 |
AChE |
4.7 |
|
115 |
eAChE |
1500 |
128 |
AChE |
6400 |
|
116 |
AChE |
89.9% inhibition |
129 |
AChE |
13800 |
|
117 |
eeAChE |
0.6 |
130 |
rAChE |
130 |
|
118 |
eeAChE |
20.97 |
131 |
eeAChE |
5570 |
|
119 |
h AChE |
142 |
132 |
eeAChE |
11 |
|
120 |
rAChE |
28 |
133 |
eeAChE |
25500 |
|
121 |
eeAChE |
53 |
134 |
eeAChE |
85 |
|
122 |
AChE |
60% inhibition |
135 |
AChE |
4580 |
|
123 |
eeAChE |
92 |
136 |
TcAChE |
160 |
|
124 |
AChE |
1500 |
137 |
rAChE |
17500 |
|
125 |
eeAChE |
720 |
138 |
AChE |
4 |
|
126 |
AChE |
13700 |
Table 7: The screening model and activity IC50 of novel heterocyclic molecules.
The 2-(2-(4-(4-Methoxyphenyl)piperazin-1-yl)acetamido)-4,5,6,7-tetrahydrobenzo [b] thiophene-3-carboxamide 122 (Figure 24) showed 60% AChE inhibition, which was stronger than donepezil (40% inhibition)in pharmacological studies at 2.6351 mM in adult male albino Wister rats [191]. Molecular modeling revealed that strong one π-π (Trp84) and three H-bond interaction [H-bond (Tyr121), H-bond (Phe331) and H-bond (Phe288)] were responsible for its potency. The piperidone grafted pyridopyrimidine-2-thione 123 (Figure 24) showed efficient AChE inhibition (IC50= 920 nM) which was 2 fold higher than galanthamine with (IC50 = 2090 nM) in Electric eel [192]. The nuciferine derivative, 124 (Figure 24) isolated from Nelumbo nucifera has been reported as a potent AChE inhibitor (IC50 = 1.5 ?M) [193,194]. It was observed that the alkoxyl group at position C-1, hydroxyl group at position C-2 and alkyl substituent at the N atom were favorable for AChE inhibition. Some pyrazoline derivatives having a dithiocarbamate moiety were reported for it AChE inhibition [195] Among them the 125 (Figure 24) bearing 2-dimethylaminoethyl and 3,4-methylenedioxyphenyl moieties showed favorable influence on the AChE inhibitory activity (IC50 = 720 nM) in Electric eel [195]. It had strong cytotoxicity (IC50 = 9.2 µM) in vitro against NIH/3T3 cells in MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay. The I-2-aryl-5-styryl-1,3,4-oxadiazole should be replaced by Among 2-aryl-5-styryl-1,3,4-oxadiazoles, the 126 (Figure 24) was also found to be a good AChE inhibitor (IC50 = 13700 nM) [196]. Among a series of 11 non-symmetrical bis quaternary pyridinium–quinolinium analogs, the 127 (Figure 24) with C-12 chain showed promising AChE inhibitory activity (IC50 = 4.7 nM) against human recombinant AChE enzyme [197]. This non-competitive type AChE inhibitor has wide scope for optimization of pharmacokinetics/pharmacological properties. The xanthine derivative propentofylline 128 (Figure 24) which is an alkaloid found to be very selective and potent AChE inhibitor (IC50 = 6400 nM) in human [198] and it was 160 fold less potent than donepezil (IC50 = 40 nM). In view of the strong anti-Aβaggregation property of benzothiazole moiety and anti-AChE activities, metal chelation, antioxidant properties and low toxicity of pyridinone moiety, these fragments linked through an appropriate spacer to provide benzothiazole–hydroxypyridinone 129 (Figure 24). This molecule inhibited eeAChE (IC50 = 13800 nM) as well as Zn induced and self-mediated-Aβ aggregation (68.9% at 100 μM) [199]. It also showed remarkable iron chelation (Zn+2 and Fe+2) andradical scavenging capacity.
 Figure 24: Heterocyclic AChE inhbitiors.
Figure 24: Heterocyclic AChE inhbitiors.
The acetophenone derivatives which possess alkylamine side chains 130 (Figure 25) showed very good AChE inhibition (IC50 = 0.13 μM) in rat cortex homogenate [200]. The benzoxazolone scaffold connected with N-substituted piperazines has been reported for AChE inhibitory activity [201,202]. Among them the analog 131 (Figure 25) exhibited strong AChE inhibition (IC50 = 0.57 pM) higher than tacrine (IC50 = 19.7 pM) and donepezil (IC50 = 57.6 pM) in Electric eel [203]. Therefore it is worth to explore this promising lead further. The 2-(4-substituted piperazine-1-yl)-N-[4-(2-methylthiazol-4-yl)phenyl] acetamide derivative 132 (Figure 25) was a significant Electric eel AChE inhibitor (IC50 = 11 nM) which was 5 fold more potent than donepezil was (IC50 = 54 nM). The thiazole derivatives 133 (Figure 25) was the very good eeAChE inhibitor (IC50 = 25500 nM) along with cytotoxic dose (IC50 = 71670 nM) of compound which was higher than its effective dose [203]. A series of triazolothiadiazoles and triazolothiadiazines has been reported for its AChE inhibitory activity. Among them the most potent derivative 134 inhibited eeAChE at nano molar rang (IC50= 65 nM) [204]. In addition it showed strong MAO-A/B inhibition (IC50 = 1.69, 32.09 µM respectively). A series of coumaryl-thiazole derivatives containing aryl urea/thiourea groups was evaluated against AChE inhibition where the most potent analog 1-(4-(8-methoxy-2-oxo-2H-chromen-3-yl)thiazol-2-yl)-3-(4-chlorophenyl) thiourea 135 (Figure 25) demonstrated remarkable eeAChE inhibitory activity (IC50 = 4580 nM) [205]. Moreover it possessed radical scavenging capacity (IC50 = 1.64 µM) against 2,2’-azino-bis(3-ethylbenzthiazoline-6-sulfonic acid (ABTS) which was significantly better than the reference quercetin (IC50 = 15.49 µM).Among the various aurone derivatives [206,207] the 3'-chloro substituted aurone derivative 136 (Figure 25) was very good AChE inhibitor (eeAChE IC50 =160 nM) [208] in Torpedo californica. It showed high passive BBB permeability and moderate CYP450 metabolic stability which indicated that this promising drug-like lead should be considering for further development. The pyrrolo-isoxazole benzoic acid derivative 137 (Figure 25) was evaluated in vitro for AChE inhibitory activity (cis, IC50 = 17.5 nM) in rat brain homogenate which was similar to donepezil (IC50 = 21.5 nM). It was also subjected to scopolamine-induced amnesia in mice at 5 and 10 mg/kg dose. The scopolamine-induced decrease attenuated in day 4 significantly (137; 49.1, doenepezil; 52.6) assessed on Morris water maze test [208]. A series of flavonoid derivatives was evaluated against AChE inhibition and most of them showed strong activity with IC50 < 100 Nm [209-213]. Among them the molecule 138 (Figure 25) was most active (IC50 = 4 nM) [209].
 Figure 25: Hetrocyclic molecules as AChE inhibitor.
Figure 25: Hetrocyclic molecules as AChE inhibitor.
MISCELLANEOUS
Apart from the above mentioned AChE inhibitors, the other potential AChE inhibitors reported in the last years are discussed below.
The phenol-berberine derivative 138 (Figure 26, Table 8) has shown mixed type very good AChE inhibitory activity (IC50 = 120 nM) in Electric eel along with excellent antioxidant activity (IC50 = 3.54 trolox equiv.) in ORAC-FL essay and anti-Aβ-aggregation capacity [214]. The bis-(-)-nor-meptazinol (bis-MEP) 139 (Figure 26) inhibited human AChE enzyme (IC50= 8640 nM), AChE-induced-Aβ aggregation (IC50 = 55300 nM) and also showed capability to complex with metal ions (Cu+2 and Zn+2) [215]. The N-[2-(diethylamino)ethyl] benzene methane sulfonamide derivative 140 (Figure 26) was evaluated against human AChE inhibition (IC50 = 2500 nM) [216]. It was predicted that the electron-withdrawing groups at para and meta positions were more favorable than the weakly electron-donating methyl group. The racemic tetrahydrocurcuminoid dihydropyrimidinone analog 141 (Figure 26) with 4-OCH3 phenyl group proved a potent human AChE inhibitor (IC50 = 1340 nM) which was more active than galanthamine (IC50= 1450 nM) [217]. Among the series of oxamide and fumaramide derivatives the analog 142 (Figure 26) was mixed-type and most potent AChE inhibition (IC50 = 30 nM) in Electric eel [218]. The phenolic derivative of symmetric sulfamide 143 (Figure 26, Table 8) showed excellent AChE inhibitory activities at nano molar concentration (IC50 = 1.96 nM) with Ki value (0.59) [219]. It exhibited significant radical scavenger capacity against DPPH (IC50 = 40760 nM), N,N-dimethyl-p-phenylenediamine radicals (DMPD, IC50 = 18230 nM) and superoxide anion radical (O2•‾), IC50 = 26200 nM). Its showed good metal chelation (Fe+2, IC50 = 18660 nM) and metal reducing power for ferric (Fe+3) and cupric (Cu+2) ions. The 1,7-diazacarbazole analog 144 (Figure 26) was a highly potent AChE inhibitor (IC50 = 11 nM) in human and potent checkpoint kinase 1 (ChK1) inhibitor [220].
 Figure 26: Miscellaneous molecules as AChE inhibitors.
Figure 26: Miscellaneous molecules as AChE inhibitors.
|
Compound No. |
Screening model |
Activity IC50 nM |
|
138 |
AChE |
4 |
|
138 |
AChE |
120 |
|
139 |
hAChE |
8640 |
|
140 |
hAChE |
2500 |
|
141 |
hAChE |
1340 |
|
142 |
eeAChE |
30 |
|
143 |
AChE |
1.96 |
|
144 |
hAChE |
11 |
Table 8: The screening model and activity IC50 of some miscellaneous.
CONCLUSION
The published clinical trial statistics indicate that in total 93 molecules have been registered under total 115 different phases of clinical trial for AD therapies till January 4, 2016. Only 24 agents reached in phase III in 36 trials. Most of the diseases modifying treatments (76%, 43% and 57% in phase III, II, I respectively) address amyloid-related targets. These statistics pointed out two things; firstly, that only a small number of molecules being assessed in clinical trial, secondly, that there is very high failure rate in advanced stage drug development for AD. It means that disease-modifying treatments alone could not provide any drug in last one decade which suggests that there was an urgent need for alternative approach. Hence this review underscores the extensive research carried out in search of AChE inhibitors as anti-alzheimer’s agents. The major approach for this has been towards the search of dual binding site AChE inhibitors as they not only bind with CAS and PAS of the enzyme but also modulated additional targets such as β-amyloid, MAO-A/B, tau inhibition, reduced the free radicals and oxidative damage. However, a large number of dual binding site AChE inhibitors reported in last decades but none of them have been proved by FDA after 2003 as an effective anti-alzheimer’s drug. That indicates that mere inhibitor design strategies based solely on SAR-conclusions are insufficient. A balanced drug profile including AChE inhibitory activity, solubility, cytotoxicity and permeability should be considered in lead optimization and further in vivo investigation. The potent inhibitors which extended beyond the previously established SARs are i) the analog 28 with an excellent AChE inhibition (IC50 = 0.006 nM) along with effective anti-Aβ(1-42) aggregation activity (52.5% at 10 µM) and anti-tau aggregation activity ( 40.7% at 10 µM) in E. coli., with good BBB permeability (Pe =10.5 (10-6 cm/s) in CNS. ii) the other example is novel templates of carbamates such as the quinoline/tetrahydroquinolin-6(-7)-yl carbamates, 56 (IC50 = 100 nM) which has shown 8 and 2 times higher therapeutic window than rivastigmine and donepezil respectively. Additionally, the later 56 showed improvement in reversible cognitive impairment induced by scopolamine as compared to existing drugs with less side effects. The other carbamate prodrug 64 acting through central cholinergic system with remarkably high AChE inhibitory activity (IC50= 20 nM) suggest that the pro drug approach could also be an alternative for rational designing of selective central AChE inhibitors.
REFERENCES
- Mount C, Downton C (2006) Alzheimer disease: progress or profit? Nat Med 12: 780-784.
- Cummings JL, Doody R, Clark C (2007) Disease-modifying therapies for Alzheimer disease: challenges to early intervention. Neurology 69: 1622-1634.
- Klafki HW, Staufenbiel M, Kornhuber J, Wiltfang J (2006) Therapeutic approaches to Alzheimer’s disease. Brain 129: 2840-2855.
- Prasher VP (2004) Review of donepezil, rivastigmine, galantamine and memantine for the treatment of dementia in Alzheimer’s disease in adults with down syndrome: implications for the intellectual disability population. Int J Geriatr Psychiatry 19: 509-515.
- Cummings JL, Morstorf T, Zhong K (2014) Alzheimer’s disease drug-development pipeline: few candidates, frequent failures. Alzheimers Res Ther 6: 37-43.
- Gerald Z, Ockert W (2013) Alzheimer’s disease market: hope deferred. Nat Rev Drug Discovery 12: 19-20.
- Anand R, Gill KD, Mahdi AA (2014) Therapeutics of Alzheimer’s disease: Past, present and future. Neuropharmacology 76: 27-50.
- Pohanka M (2012) Acetylcholinesterase inhibitors: a patent review (2008 - present). Expert Opin Ther Pat 22: 871-886.
- Francis PT, Palmer AM, Snape M, Wilcock GK (1999) The cholinergic hypothesis of Alzheimer’s disease: a review of progress. J Neurol Neurosurg Psychiatry 66: 137-147.
- Sussman JL, Harel M, Silman I (1993) Three-dimensional structure of acetylcholinesterase and of its complexes with anticholinesterase drugs. Chem Biol Interact 87: 187-197.
- Nachon F, Carletti E, Ronco C, Trovaslet M, Nicolet Y, et al. (2013) Crystal structures of human cholinesterases in complex with huprine W and tacrine: elements of specificity for anti-Alzheimer’s drugs targeting acetyl- and butyryl-cholinesterase. Biochem J 453: 393-399.
- Inestrosa NC, Alvarez A, Perez CA, Moreno RD, Vicente M, et al. (1996) Acetylcholinesterase accelerates assembly of amyloid-beta-peptides into Alzheimer’s fibrils: possible role of the peripheral site of the enzyme. Neuron 16: 881-891.
- Alvarez A, Opazo C, Alarcon R, Garrido J, Inestrosa NC (1997) Acetylcholinesterase promotes the aggregation of amyloid-beta-peptide fragments by forming a complex with the growing fibrils. J Mol Biol 272: 348-361.
- De Ferrari GV, Canales MA, Shin I, Weiner LM, Silman I, et al. (2001) A structural motif of acetylcholinesterase that promotes amyloid beta-peptide fibril formation. Biochemistry 40: 10447-10457.
- Diamant S, Podoly E, Friedler A, Ligumsky H, Livnah O, et al. (2006) Butyrylcholinesterase attenuates amyloid fibril formation in vitro. Proc Natl Acad Sci USA 103: 8628-8633.
- Podoly E, Bruck T, Diamant S, Melamed-Book N, Weiss A, (2008) Human recombinant butyrylcholinesterase purified from the milk of transgenic goats interacts with beta-amyloid fibrils and suppresses their formation in vitro. Neurodegener Dis 5: 232-236.
- Watkins PB, Zimmerman HJ, Knapp MJ, Gracon SI, Lewis KW (1994) hepatotoxic effects of tacrine administration in patients with Alzheimer’s disease. JAMA 271: 992-998.
- Bajda M, Guzior N, Ignasik M, Malawska B (2011) Multi-target-directed ligands in Alzheimer’s disease treatment. Curr Med Chem 18: 4949-4975.
- Rampa A, Belluti F, Gobbi S, Bisi A (2011) Hybrid-based multi-target ligands for the treatment of Alzheimer’s disease. Curr Top Med Chem 11: 2716-2730.
- de los Ríos C (2012) Cholinesterase inhibitors: a patent review (2007 - 2011). Expert Opin Ther Pat 22: 853-869.
- Anand P, Singh B (2013) A review on cholinesterase inhibitors for Alzheimer’s disease. Arch Pharm Res 36: 375-399.
- Guzior N, Wieckowska A, Panek D, Malawska B (2015) Recent development of multifunctional agents as potential drug candidates for the treatment of Alzheimer’s disease. Curr Med Chem 22: 373-404.
- Singh M, Kaur M, Chadha N, Silakari O (2016) Hybrids: a new paradigm to treat Alzheimer’s disease. Mol Divers 20: 271-297.
- Matias M, Silvestre S, Falcao A, Alves G (2017) Recent Highlights on Molecular Hybrids Potentially Useful in Central Nervous System Disorders. Mini Rev Med Chem 17: 486-517.
- Wlodek ST, Antosiewicz J, McCammon JA, Straatsma TP, Gilson MK, et al. (1996) Binding of tacrine and 6-chlorotacrine by acetylcholinesterase. Biopolymers 38: 109-117.
- Korabecny J, Janovec L, Musilek K, Zemek F, Horova A, et al. (2013) Comparison of novel tacrine and 7-MEOTA derivatives with aromatic and alicyclic residues: synthesis, biological evaluation and docking studies. Lett Org Chem 10: 291-297.
- Soukup O, Jun D, Zdarova-Karasova J, Patocka J, Musilek K, et al. (2013) A resurrection of 7-MEOTA: a comparison with Tacrine. Curr Alzheimer Res 10: 893-906.
- Kozurkova M, Hamulakova S, Gazova Z, Paulikova H, Kristian P (2011) Neuroactive multifunctional tacrine congeners with cholinesterase, anti-amyloid aggregation and neuroprotective properties. Pharmaceuticals 4: 382-418.
- Minarini A, Milelli A, Simoni E, Rosini M, Bolognesi ML, et al. (2013) Multifunctional tacrine derivatives in Alzheimer’s disease. Curr Top Med Chem 13: 1771-1786.
- Pang Y-P, Brimijoin S (1997) Tha analogs useful as cholinesterase inhibitors. Patent WO1997021681, Mayo Foundation For Medical Education And Research, USA.
- Lin H, Li Q, Gu K, Zhu J, Jiang X, et al. (2017) Therapeutic Agents in Alzheimer’s Disease Through a Multi-targetdirected Ligands Strategy: Recent Progress Based on Tacrine Core. Curr Top Med Chem 17: 3000-3016.
- Sameem B, Saeedi M, Mahdavi M, Shafiee A (2017) A review on tacrine-based scaffolds as multi-target drugs (MTDLs) for Alzheimer’s disease. Eur J Med Chem 128: 332-345.
- Wu WY, Dai YC, Li NG, Dong ZX, Gu T, et al. (2017) Novel multitarget-directed tacrine derivatives as potential candidates for the treatment of Alzheimer’s disease. J Enzyme Inhib Med Chem 32: 572-587.
- Ismaili L, Refouvelet B, Benchekroun M, Brogi S, Brindisi M, et al. (2017) Multitarget compounds bearing tacrine- and donepezil-like structural and functional motifs for the potential treatment of Alzheimer’s disease. Prog Neurobiol 151: 4-34.
- Hamulakova S, Janovec L, Hrabinova M, Kristian P, Kuca K, et al. (2012) Synthesis, design and biological evaluation of novel highly potent tacrine congeners for the treatment of Alzheimer’s disease. Eur J Med Chem 55: 23-31.
- Hamulakova S, Imrich J, Janovec L, Kristian P, Danihel I, et al. (2014) Novel tacrine/acridine anticholinesterase inhibitors with piperazine and thiourea linkers. Int J Biol Macromol 70: 435-439.
- Qian S, He L, Mak M, Han Y, Ho CY, et al. (2014) Synthesis, biological activity, and biopharmaceutical characterization of tacrine dimers as acetylcholinesterase inhibitors. Int J Pharm 477: 442-453.
- Luo W, Li YP, He Y, Huang SL, Tan JH, et al. (2011) Design, synthesis and evaluation of novel tacrine-multialkoxybenzene hybrids as dual inhibitors for cholinesterases and amyloid beta aggregation. Bioorg Med Chem 19: 763-770.
- Luo W, Li YP, He Y, Huang SL, Li D, et al. (2011) Synthesis and evaluation of heterobivalent tacrine derivatives as potential multi-functional anti-Alzheimer agents. Eur J Med Chem 46: 2609-2616.
- Szymanski P, Karpinski A, Mikiciuk-Olasik E (2011) Synthesis, biological activity and HPLC validation of 1,2,3,4-tetrahydroacridine derivatives as acetylcholinesterase inhibitors. Eur J Med Chem 46: 3250-3257.
- Szymanski P, Markowicz M, Mikiciuk-Olasik E (2011) Synthesis and biological activity of derivatives of tetrahydroacridine as acetylcholinesterase inhibitors. Bioorg Chem 39: 138-142.
- Szymanski P, Laznickova A, Laznicek M, Bajda M, Malawska B, et al. (2012) 2,3-Dihydro-1H-cyclopenta[b]quinoline derivatives as acetylcholinesterase inhibitors-synthesis, radiolabeling and biodistribution. Int J Mol Sci 13: 10067-10090.
- Badran MM, Hakeem MA, Abuel-Maaty SM, El-Malah A, Salam RMA (2012) Design and synthesis of thienopyridines as novel templates for acetylcholinesterase inhibitors. Med Chem Res 22: 4087-4095.
- Huang L, Su T, Shan W, Luo Z, Sun Y, et al. (2012) Inhibition of cholinesterase activity and amyloid aggregation by berberine-phenyl-benzoheterocyclic and tacrine-phenyl-benzoheterocyclic hybrids. Bioorg Med Chem 20: 3038-3048.
- Keri RS, Quintanova C, Marques SM, Esteves AR, Cardoso SM, et al. (2013) Design, synthesis and neuroprotective evaluation of novel tacrine-benzothiazole hybrids as multi-targeted compounds against Alzheimer’s disease. Bioorg Med Chem 21: 4559-4569.
- Wang Y, Guan XL, Wu PF, Wang CM, Cao H, et al. (2012) Multifunctional mercapto-tacrine derivatives for treatment of age-related neurodegenerative diseases. J Med Chem 55: 3588-3592.
- Chen Y, Sun J, Fang L, Liu M, Peng S, et al. (2012) Tacrine-ferulic acid-nitric oxide (NO) donor trihybrids as potent, multifunctional acetyl- and butyrylcholinesterase inhibitors. J Med Chem 55: 4309-4321.
- Chen Y, Sun J, Huang Z, Liao H, Peng S, et al. (2013) Design, synthesis and evaluation of tacrine-flurbiprofen-nitrate trihybrids as novel anti-Alzheimer’s disease agents. Bioorg Med Chem 21: 2462-2470.
- da Costa JS, Lopes JP, Russowsky D, Petzhold CL, Borges AC, et al. (2013) Synthesis of tacrine-lophine hybrids via one-pot four component reaction and biological evaluation as acetyl- and butyrylcholinesterase inhibitors. Eur J Med Chem 62: 556-563.
- Kochi A, Eckroat TJ, Green KD, Mayhoub AS, Lim MH, et al. (2013) A novel hybrid of 6-chlorotacrine and metal-amyloid-β modulator for inhibition of acetylcholinesterase and metal-induced amyloid-β Chem Sci 4: 4137-4145.
- Fernandez-Bachiller MI, Perez C, Monjas L, Rademann J, Rodriguez-Franco MI (2012) New tacrine-4-oxo-4H-chromene hybrids as multifunctional agents for the treatment of Alzheimer’s disease, with cholinergic, antioxidant, and beta-amyloid-reducing properties. J Med Chem 55: 1303-1317.
- Liao S, Deng H, Huang S, Yang J, Wang S, et al. (2015) Design, synthesis and evaluation of novel 5,6,7-trimethoxyflavone-6-chlorotacrine hybrids as potential multifunctional agents for the treatment of Alzheimer’s disease. Bioorg Med Chem Lett 25: 1541-1545.
- Li S-Y, Wang X-B, Xie S-S, Jiang N, Wang KD, et al. (2013) Multifunctional tacrine-flavonoid hybrids with cholinergic, β-amyloid-reducing, and metal chelating properties for the treatment of Alzheimer’s disease. Eur J Med Chem 69: 632-646.
- Xie S-S, Wang X-B, Li J-Y, Yang L, Kong L-Y. (2013) Design, synthesis and evaluation of novel tacrine-coumarin hybrids as multifunctional cholinesterase inhibitors against Alzheimer’s disease. Eur J Med Chem 64: 540-553.
- Xie S-S, Wang X, Jiang N, Yu W, Wang KD, et al. (2015) Multi-target tacrine-coumarin hybrids: Cholinesterase and monoamine oxidase-B inhibition properties against Alzheimer’s disease. Eur J Med Chem 95: 153-165.
- Hamulakova S, Janovec L, Hrabinova M, Spilovska K, Korabecny J, et al. (2014) Synthesis and biological evaluation of novel tacrine derivatives and tacrine-coumarin hybrids as cholinesterase inhibitors. J Med Chem 57: 7073-7084.
- Sun Q, Peng DY, Yang SG, Zhu XL, Yang WC, et al. (2014) Syntheses of coumarin-tacrine hybrids as dual-site acetylcholinesterase inhibitors and their activity against butylcholinesterase, Aβ aggregation, and β-secretase. Bioorg Med Chem 22: 4784-4791.
- Khoobi M, Alipour M, Moradi A, Sakhteman A, Nadri H, et al. (2013) Design, synthesis, docking study and biological evaluation of some novel tetrahydrochromeno[3′,4′:5,6]pyrano[2,3-b]quinolin-6(7H)-one derivatives against acetyl- and butyrylcholinesterase. Eur J Med Chem 68: 291-300.
- Spilovska K, Korabecny J, Kral J, Horova A, Musilek K, et al. (2013) 7-Methoxytacrine-adamantylamine heterodimers as cholinesterase inhibitors in Alzheimer’s disease treatment-synthesis, biological evaluation and molecular modeling studies. Molecules 18: 2397-2418.
- Korabecny J, Andrs M, Nepovimova E, Dolezal R, Babkova K, et al. (2015) 7-Methoxytacrine-p-anisidine hybrids as novel dual binding site acetylcholinesterase inhibitors for Alzheimer's disease treatment. Molecules 20: 22084-22101.
- Lu C, Zhou Q, Yan J, Du Z, Huang L, et al. (2013) A novel series of tacrine-selegiline hybrids with cholinesterase and monoamine oxidase inhibition activities for the treatment of Alzheimer’s disease. Eur J Med Chem 62: 745-753.
- Zawadzka A, Lozinska I, Moleda Z, Panasiewicz M, Czarnocki Z (2013) Highly selective inhibition of butyrylcholinesterase by a novel melatonin-tacrine heterodimers. J Pineal Res 54: 435-441.
- Di Pietro O, Pérez-Areales FJ, Juárez-Jiménez J, Espargaró A, Clos MV, et al. (2014) Tetrahydrobenzo[h][1,6]naphthyridine-6-chlorotacrine hybrids as a new family of anti-Alzheimer agents targeting β-amyloid, tau, and cholinesterase pathologies. Eur J Med Chem 84: 107-117.
- El-Malah A, Gedawy EM, Kassab AE, Salam RM (2014) Novel tacrine analogs as potential cholinesterase inhibitors in Alzheimer’s disease. Arch Pharm (Weinheim) 347: 96-103.
- Hui A-L, Chen Y, Zhu S-J, Gan C-S, Pan J, et al. (2014) Design and synthesis of tacrine-phenothiazine hybrids as multitarget drugs for Alzheimer’s disease. Med Chem Res 23: 3546-3557.
- Ling H, Jian P, Sheng C (2015) A tacrine-phenothiazine iso-diastereomer compound and a preparation method thereof. Patent CN102936244 B, Hefei University of Technology, Hefei, China.
- Kalai T, Altman R, Maezawa I, Balog M, Morisseau C, et al. (2014) Synthesis and functional survey of new tacrine analogs modified with nitroxides or their precursors. Eur J Med Chem 77: 343-350.
- Lan JS, Xie SS, Li SY, Pan LF, Wang XB, et al. (2014) Design, synthesis and evaluation of novel tacrine-(β-carboline) hybrids as multifunctional agents for the treatment of Alzheimer’s disease. Bioorg Med Chem 22: 6089-6104.
- Nepovimova E, Uliassi E, Korabecny J, Peña-Altamira LE, Samez S, et al. (2014) Multitarget drug design strategy: quinone–tacrine hybrids designed to block amyloid-β aggregation and to exert anticholinesterase and antioxidant effects. J Med Chem 57: 8576-8589.
- Bajda M, Jonczyk J, Malawska B, Czarnecka K, Girek M, et al. (2015) Synthesis, biological evaluation and molecular modeling of new tetrahydroacridine derivatives as potential multifunctional agents for the treatment of Alzheimer's disease. Bioorg Med Chem 23: 5610-5618.
- Chao X, He X, Yang Y, Zhou X, Jin M, et al. (2012) Design, synthesis and pharmacological evaluation of novel tacrine–caffeic acid hybrids as multi-targeted compounds against Alzheimer’s disease. Bioorg Med Chem Lett 22: 6498-6502.
- Pei-qing L, Jiantao Y, Xixin he, Cheng Z, Yang Y, et al. (2012) Tacrine-caffeic acid hetero-blends, their preparation method and medicinal compositions. Patent CN102617465, Nakayama University, Japan.
- Digiacomo M, Chen Z, Wang S, Lapucci A, Macchia M, et al. (2015) Synthesis and pharmacological evaluation of multifunctional tacrine derivatives against several disease pathways of AD. Bioorg Med Chem Lett 25: 807-810.
- Benchekroun M, Bartolini M, Egea J, Romero A, Soriano E, et al. (2015) Novel tacrine-grafted ugi adducts as multipotent anti-Alzheimer drugs: a synthetic renewal in tacrine-ferulic acid hybrids. Chem Med Chem 10: 523-539.
- Mohammadi-Khanaposhtani M, Saeedi M, Zafarghandi NS, Mahdavi M, Sabourian R, et al. (2015) Potent acetylcholinesterase inhibitors: design, synthesis, biological evaluation, and docking study of acridone linked to 1,2,3-triazole derivatives. Eur J Med Chem 92: 799-806.
- Nepovimova E, Korabecny J, Dolezal R, Babkova K, Ondrejicek A, et al. (2015) Tacrine–trolox hybrids: A novel class of centrally active, nonhepatotoxic multi-target-directed ligands exerting anticholinesterase and antioxidant activities with low in vivo J Med Chem 58: 8985-9003.
- Xie S-S, Lan J-S, Wang X-B, Jiang N, Dong G, et al. (2015) Multifunctional tacrine-trolox hybrids for the treatment of Alzheimer’s disease with cholinergic, antioxidant, neuroprotective and hepatoprotective properties. Eur J Med Chem 93: 42-50.
- Rong C, Li DL, Sen Y, Lin F (2014) Hydrogenated acridine derivative and application thereof. Patent CN103524413, Jiangsu Simcere Pharmaceutical Co., Ltd., China.
- Rong C, Fei L, Xin C, Lin F, Island F, et al. (2012) Compounds and uses as inhibitors of I-type calcium channel blockers and/or acetylcholinesterase. Patent WO2012065527, Jiangsu Xiangyao Drug Research Co., Ltd., Jiangsu, China.
- De LRSC, Romero MA, Egea MJ, Leon MR, Villarroya SM, et al. (2011). Compounds derived from 1,8-naphthyridines and their use in the treatment of neurodegenerative diseases. Patent WO2011051527A1, Higher Council for Scientific Research CSIC, Spain.
- Leon R, Marco-Contelles J. (2011) A step further towards multitarget drugs for Alzheimer and neuronal vascular diseases: targeting the cholinergic system, amyloid-β aggregation and Ca2+ Curr Med Chem 18: 552-576.
- Silva D, Chioua M, Samadi A, Carmo Carreiras M, Jimeno ML, et al. (2011) Synthesis and pharmacological assessment of diversely substituted pyrazolo[3,4-b]quinoline, and benzo[b]pyrazolo[4,3-g][1,8]naphthyridine derivatives. Eur J Med Chem 46: 4676-4681.
- Chioua M, Perez M, Bautista-Aguilera OM, Yanez M, Lopez MG, et al. (2015) Development of huper tacrines as non-toxic, cholinesterase inhibitors for the potential treatment of Alzheimer’s disease. Mini-Rev Med Chem 15: 648-658.
- Chen R, Feng L, Zhang Z, Fang F, Dong Q, et al. (2014) 1,4-Dihydro-naphthyridine derivative and pharmaceutical composition and use thereof. Patent US20140364443A, Jiangsu Simovay Pharmaceutical Co., Ltd., China.
- Wang XL, Xiong Y, Yang Y, Tuo QZ, Wang XC, et al. (2015) A novel tacrine-dihydropyridine hybrid (-)SCR1693 induces tau dephosphorylation and inhibits Aβ generation in cells. Eur J Pharmacol 754: 134-139.
- Zhang Z, Chen R, An W, Wang C, Liao G, et al. (2016) A novel acetylcholinesterase inhibitor and calcium channel blocker SCR-1693 improves Aβ25–35-impaired mouse cognitive function. Psychopharmacology (Berl) 233: 599-613.
- Chen R, Zhengping Z, Lin F, Dong Q, Fulong L, et al. (2013) 5-amino-1,4-dihydro-1,8-naphthyridine derivative and pharmaceutical composition and use thereof. Patent WO2013152704A1, Jiangsu Xianxian Pharmaceutical Co., Ltd., Shanghai, China.
- Samadi A, de los Rios C, Bolea I, Chioua M, Iriepa I, et al. (2012) Multipotent MAO and cholinesterase inhibitors for the treatment of Alzheimer’s disease: Synthesis, pharmacological analysis and molecular modeling of heterocyclic substituted alkyl and cycloalkyl propargyl amine. Eur J Med Chem 52: 251-262.
- Onor ML, Trevisiol M, Aguglia E (2007) Rivastigmine in the treatment of Alzheimer’s disease: an update. Clin Interv Aging 2: 17-32.
- Zheng H, Youdim MBH, Fridkin M (2009) Site-activated multifunctional chelator with acetylcholinesterase and neuroprotective−neurorestorative moieties for alzheimer’s therapy. J Med Chem 52: 4095-4098.
- Youdim MBH, Fridkin M, Zheng H (2014) Neuroprotective multifunctional compounds and pharmaceutical compositions comprising them. Patent US8802675 B2. Technion Research And Development Foundation Ltd., Yeda Research And Development Company Ltd., Israel.
- Zheng H, Amit T, Bar-Am O, Fridkin M, Youdim MB, et al. (2012) From anti-Parkinson’s drug rasagiline to novel multitarget iron chelators with acetylcholinesterase and monoamine oxidase inhibitory and neuroprotective properties for Alzheimer’s disease. J Alzheimers Dis 30: 1-16.
- Zheng H, Youdim MB, Fridkin M (2010) Selective acetylcholinesterase inhibitor activated by acetylcholinesterase releases an active chelator with neurorescuing and anti-amyloid activities. ACS Chem Neurosci 1: 737-746.
- Zheng H, Youdim MB, Fridkin M (2010) Site-activated chelators targeting acetylcholinesterase and monoamine oxidase for Alzheimer's therapy. ACS Chem Biol 5: 603-610.
- Wang L, Esteban G, Ojima M, Bautista-Aguilera OM, Inokuchi T, et al. (2014) Donepezil + propargylamine + 8-hydroxyquinoline hybrids as new multifunctional metal-chelators, ChE and MAO inhibitors for the potential treatment of Alzheimer’s disease. Eur J Med Chem 80: 543-561.
- Long Y, Zhang H, Zhou T, Liu Z, Zou Z (2013) 2-substutied-3-quinolinic acid ethyl ester compound, preparation method and applications thereof . Patent CN102584698B, South China Normal University, China.
- Wenhai H , Zhengrong C, Ye Y, Zhen M, yuan Z (2014) 6,8-disubstituted quinoline compound or pharmaceutically acceptable salt thereof, preparing method and application of same. Patent CN103980194, Zhejiang Academy of Medical Sciences, China.
- Shakya N, Fatima Z, Nath C, Saxena AK (2006) Substituted carbamic acid quinolin-6-yl esters useful as acetylcholinesterase inhibitors. (patent WO2006070394), Council Of Scientific And Industrial Research, Hyderabad, India.
- Chaudhaery SS, Roy KK, Shakya N, Saxena G, Sammi SR, et al. (2010) Novel carbamates as orally active acetylcholinesterase inhibitors found to improve scopolamine-induced cognition impairment: Pharmacophore-based virtual screening, synthesis, and pharmacology. J Med Chem 53: 6490-6505.
- Roy KK, Tota S, Tripathi T, Chander S, Nath C, et al. (2012) Lead optimization studies towards the discovery of novel carbamates as potent AChE inhibitors for the potential treatment of Alzheimer’s disease. Bioorg Med Chem 20: 6313-6320.
- Roy KK, Tota S, Nath C, Shukla R, Saxena AK (2013) Substituted 1, 2, 3, 4-tetrahydroquinolin-7-yl carbamates as acetylcholinesterase inhibitors for treatment of alzheimer’s disease. Patent US20130324573A1, Council of Scientific & Industrial Research (CSIR), India.
- Sen Y, Lin F, Qingli D, Rong C (2014) Piperazine compound and its application. Patent CN102603675B, Jiangsu Xiangyao Drug Research Co., Ltd., Jiangsu, China.
- Imramovsky A, Stepankova S, Vanco J, Pauk K, Monreal-Ferriz J, et al. (2012) Acetylcholinesterase-inhibiting activity of salicylanilide N-alkylcarbamates and their molecular docking. Molecules 17: 10142-10158.
- Anand P, Singh B (2013) Synthesis and evaluation of substituted 4-methyl-2-oxo-2H-chromen-7-yl phenyl carbamates as potent acetylcholinesterase inhibitors and anti-amnestic agents. Med Chem 9: 694-702.
- Shao BY, Xia Z, Xie Q, Ge XX, Zhang WW, et al. (2014) Meserine, a novel carbamate AChE inhibitor, ameliorates scopolamine-induced dementia and alleviates amyloidogenesis of APP/PS1 transgenic mice. CNS Neurosci Ther 20: 165-171.
- Bohn P, Gourand F, Papamicael C, Ibazizene M, Dhilly M, et al. (2015) Dihydroquinoline Carbamate Derivatives as “Bio-oxidizable” Prodrugs for Brain Delivery of Acetylcholinesterase Inhibitors: [¹¹C] Radiosynthesis and Biological Evaluation. ACS Chem Neurosci 6: 737-744.
- Doody RS, Cummings JL, Farlow MR (2012) Reviewing the role of donepezil in the treatment of Alzheimer’s disease. Curr Alzheimer Res 9: 773-781.
- Rodrigues Simoes MC, Dias Viegas FP, Moreira MS, de Freitas Silva M, Riquiel MM, et al. (2014) Donepezil: an important prototype to the design of new drug candidates for Alzheimer’s disease. Mini Rev Med Chem 14: 2-19.
- Gant TG, Sarshar S, Shahbaz MM (2010) Indanone inhibitors of acetylcholinesterase. US20100143505A1, Auspex Pharmaceuticals, Inc, California, USA.
- Belluti F, Bartolini M, Bottegoni G, Bisi A, Cavalli A, et al. (2011) Benzophenone-based derivatives: a novel series of potent and selective dual inhibitors of acetylcholinesterase and acetylcholinesterase-induced beta-amyloid aggregation. Eur J Med Chem 46: 1682-1693.
- Bolea I, Juárez-Jiménez J, de Los Ríos C, Chioua M, Pouplana R, et al. (2011) Synthesis, biological evaluation, and molecular modeling of donepezil and N-[(5-(Benzyloxy)-1-methyl-1H-indol-2-yl)methyl]-N-methylprop-2-yn-1-amine hybrids as new multipotent cholinesterase/monoamine oxidase inhibitors for the treatment of Alzheimer’s disease. J Med Chem 54: 8251-8270.
- Esteban G, Allan J, Samadi A, Mattevi A, Unzeta M, et al. (2014) Kinetic and structural analysis of the irreversible inhibition of human monoamine oxidases by ASS234, a multi-target compound designed for use in Alzheimer’s disease. Biochim Biophys Acta 1844: 1104-1110.
- Bautista-Aguilera OM, Esteban G, Bolea I, Nikolic K, Agbaba D, et al. (2014) Design, synthesis, pharmacological evaluation, QSAR analysis, molecular modeling and ADMET of novel donepezil-indolyl hybrids as multipotent cholinesterase/monoamine oxidase inhibitors for the potential treatment of Alzheimer’s disease. Eur J Med Chem 75: 82-95.
- Bolea I, Gella A, Monjas L, Pérez C, Rodríguez-Franco M, et al. (2013) Multipotent, permeable drug ASS234 inhibits Aβ aggregation, possesses antioxidant properties and protects from Aβ-induced apoptosis in vitro. Curr Alzheimer Res 10: 797-808.
- Samadi A, Estrada M, Perez C, Rodriguez-Franco MI, Iriepa I, et al. (2012) Pyridonepezils, new dual AChE inhibitors as potential drugs for the treatment of Alzheimer’s disease: synthesis, biological assessment, and molecular modeling. Eur J Med Chem 57: 296-301.
- Samadi A, Fuente-Revenga M, Perez C, Iriepa I, Moraleda I, et al. (2013) Synthesis, pharmacological assessment, and molecular modeling of 6-chloro-pyridonepezils: New dual AChE inhibitors as potential drugs for the treatment of Alzheimer’s disease. Eur J Med Chem 67: 64-74.
- Meng FC, Mao F, Shan WJ, Qin F, Huang L, et al. (2012) Design, synthesis, and evaluation of indanone derivatives as acetylcholinesterase inhibitors and metal-chelating agents. Bioorg Med Chem Lett 22: 4462-4466.
- Huang L, Miao H, Sun Y, Meng F, Li X (2014) Discovery of indanone derivatives as multi-target-directed ligands against Alzheimer’s disease. Eur J Med Chem 87: 429-439.
- Asadipour A, Alipour M, Jafari M, Khoobi M, Emami S, et al. (2013) Novel coumarin-3-carboxamides bearing N-benzylpiperidine moiety as potent acetylcholinesterase inhibitors. Eur J Med Chem 70: 623-630.
- Modh RP, Kumar SP, Jasrai YT, Chikhalia KH (2013) Design, synthesis, biological evaluation, and molecular modeling of coumarin-piperazine derivatives as acetylcholinesterase inhibitors. Arch Pharm (Weinheim) 346: 793-804.
- Razavi SF, Khoobi M, Nadri H, Sakhteman A, Moradi A, et al. (2013) Synthesis and evaluation of 4-substituted coumarins as novel acetylcholinesterase inhibitors. Eur J Med Chem 64: 252-259.
- Alipour M, Khoobi M, Moradi A, Nadri H, Moghadam FH, et al. (2014) Synthesis and anti-cholinesterase activity of new 7-hydroxycoumarin derivatives. Eur J Med Chem 82: 536-544.
- Tarozzi A, Bartolini M, Piazzi L, Valgimigli L, Amorati R, et al. (2014) From the dual function lead AP2238 to AP2469, a multi-target-directed ligand for the treatment of Alzheimer’s disease. Pharmacol Res Perspect 2: 23.
- Farina R, Pisani L, Catto M, Nicolotti O, Gadaleta D, et al. (2015) Structure-based design and optimization of multitarget-directed 2H-chromen-2-one derivatives as potent inhibitors of monoamine oxidase b and cholinesterases. J Med Chem 58: 5561-5578.
- Luo Z, Sheng J, Sun Y, Lu C, Yan J, et al. (2013) Synthesis and evaluation of multi-target-directed ligands against Alzheimer’s disease based on the fusion of donepezil and ebselen. J Med Chem 56: 9089-9099.
- Nadri H, Pirali-Hamedani M, Moradi A, Sakhteman A, Vahidi A, et al. (2013) 5,6-Dimethoxybenzofuran-3-one derivatives: a novel series of dual Acetylcholinesterase/Butyrylcholinesterase inhibitors bearing benzyl pyridinium moiety. Daru 21: 15.
- Baharloo F, Moslemin MH, Nadri H, Asadipour A, Mahdavi M, et al. (2015) Benzofuran-derived benzylpyridinium bromides as potent acetylcholinesterase inhibitors. Eur J Med Chem 93: 196-201.
- Arab S, Sadat-Ebrahimi SE, Mohammadi-Khanaposhtani M, Moradi A, Nadri H, et al. (2015) Synthesis and Evaluation of Chroman-4-One Linked to N-Benzyl Pyridinium Derivatives as New Acetylcholinesterase Inhibitors. Arch Pharm (Weinheim) 348: 643-649.
- Wang C, Wu Z, Cai H, Xu S, Liu J, et al. (2015) Design, synthesis, biological evaluation and docking study of 4-isochromanone hybrids bearing N-benzyl pyridinium moiety as dual binding site acetylcholinesterase inhibitors. Bioorg Med Chem Lett 25: 5212-5216.
- Akrami H, Mirjalili BF, Khoobi M, Nadri H, Moradi A, et al. (2014) Indolinone-based acetylcholinesterase inhibitors: Synthesis, biological activity and molecular modeling. Eur J Med Chem 84: 375-381.
- Mostofi M, Mohammadi Ziarani G, Mahdavi M, Moradi A, Nadri H, et al. (2015) Synthesis and structure-activity relationship study of benzofuran-based chalconoids bearing benzylpyridinium moiety as potent acetylcholinesterase inhibitors. Eur J Med Chem 103: 361-369.
- Ismail MM, Kamel MM, Mohamed LW, Faggal SI (2012) Synthesis of new indole derivatives structurally related to donepezil and their biological evaluation as acetylcholinesterase inhibitors. Molecules 17: 4811-4823.
- Mohammadi-Farani A, Ahmadi A, Nadri H, Aliabadi A (2013) Synthesis, docking and acetylcholinesterase inhibitory assessment of 2-(2-(4-Benzylpiperazin-1-yl)ethyl)isoindoline-1,3-dione derivatives with potential anti-Alzheimer effects. Daru 21: 47.
- Guzior N, Bajda M, Rakoczy J, Brus B, Gobec S, et al. (2015) Isoindoline-1,3-dione derivatives targeting cholinesterases: Design, synthesis and biological evaluation of potential anti-Alzheimer’s agents. Bioorg Med Chem 23: 1629-1637.
- Guzior N, Bajda M, Skrok M, Kurpiewska K, Lewinski K, et al. (2015) Development of multifunctional, heterodimeric isoindoline-1,3-dione derivatives as cholinesterase and β-amyloid aggregation inhibitors with neuroprotective properties. Eur J Med Chem 92: 738-749.
- Rayatzadeh A, Saeedi M, Mahdavi M, Rezaei Z, Sabourian R, et al. (2015) Synthesis and evaluation of novel oxoisoindoline derivatives as acetylcholinesterase inhibitors. Monatsh Chem 146: 637.
- Zurek E, Szymanski P, Mikiciuk-Olasik E (2013) Synthesis and biological activity of new donepezil-hydrazinonicotinamide hybrids. Drug Res (Stuttg) 63: 137-144.
- Ma X, Gang DR (2004) The Lycopodium alkaloids. Nat Prod Rep 21: 752-772.
- Qian ZM, Ke Y (2014) Huperzine A: Is it an effective disease-modifying drug for Alzheimer’s disease?. Front Aging Neurosci 6: 216.
- Zhang HY (2012) New insights into huperzine A for the treatment of Alzheimer’s disease. Acta Pharmacol Sin 33: 1170-1175.
- Rafii MS, Walsh S, Little JT, Behan K, Reynolds B, et al. (2011) A phase II trial of huperzine A in mild to moderate Alzheimer disease. Neurology 76: 1389-1394.
- Jia JY, Zhao QH, Liu Y, Gui YZ, Liu GY, et al. (2013) Phase I study on the pharmacokinetics and tolerance of ZT-1, a prodrug of huperzine A, for the treatment of Alzheimer’s disease. Acta Pharmacol Sin 34: 976-982.
- Yang G, Wang Y, Tian J, Liu J-P (2013) Huperzine A for Azheimer’s disease: a systematic review and meta-analysis of randomized clinical trials. PLoS One 8: 74916.
- Munoz-Torrero D, Camps P (2008) Huprines for Alzheimer’s disease drug development. Expert Opin Drug Discov 3: 65-81.
- Ratia M, Gimenez-Llort L, Camps P, Munoz-Torrero D, Perez B, et al. (2013) Huprine X and huperzine A improve cognition and regulate some neurochemical processes related with Alzheimer’s disease in triple transgenic mice (3xTg-AD). Neurodegener Dis 11: 129-140.
- Ronco C, Renard PY, Jean L, Nachon F, Romieu A (2011) 7,11-Methanocycloocta [b] quinoline derivative as highly functionalizable acetylcholinesterase inhibitors. Universite De Rouen, Mont-Saint-Aignan, France.
- Ronco C, Foucault R, Gillon E, Bohn P, Nachon F, et al. (2011) New huprine derivatives functionalized at position 9 as highly potent acetylcholinesterase inhibitors. Chem Med Chem 6: 876-888.
- Ronco C, Carletti E, Colletier JP, Weik M, Nachon F, et al. (2012) Huprine derivatives as sub-nanomolar human acetylcholinesterase inhibitors: from rational design to validation by X-ray crystallography. Chem Med Chem 7: 400-405.
- Oueis E, Santoni G, Ronco C, Syzgantseva O, Tognetti V, et al. (2014) Reaction site-driven regioselective synthesis of AChE inhibitors. Org Biomol Chem 12: 156-161.
- Viayna E, Sola I, Bartolini M, De Simone A, Tapia-Rojas C, et al. (2014) Synthesis and multitarget biological profiling of a novel family of rhein derivatives as disease-modifying anti-Alzheimer agents. J Med Chem 57: 2549-2567.
- Sola I, Artigas A, Taylor MC, Gbedema SY, Pérez B, et al. (2014) Synthesis and antiprotozoal activity of oligomethylene- and p-phenylene-bis(methylene)-linked bis(+)-huprines. Bioorg Med Chem Lett 24: 5435-5438.
- Pérez-Areales FJ, Di Pietro O, Espargaró A, Vallverdú-Queralt A, Galdeano C, et al. (2014) Shogaol-huprine hybrids: dual antioxidant and anticholinesterase agents with β-amyloid and tau anti-aggregating properties. Bioorg Med Chem 22: 5298-5307.
- Sola I, Aso E, Frattini D, López-González I, Espargaró A, et al. (2015) Novel levetiracetam derivatives that are effective against the Alzheimer-like phenotype in mice: Synthesis, in vitro, ex vivo, and in vivo efficacy studies. J Med Chem 58: 6018-6032.
- Muñoz-torrero L-ID, Ferrer AI, Sola LI, Aso PE (2014) Multi-target drug compounds for the treatment of neurodegenerative disorders. (patent WO2014206877), Universitat De Barcelona, Barcelona, Spain.
- Galdeano C, Viayna E, Sola I, Formosa X, Camps P, et al. (2012) Huprine-tacrine heterodimers as anti-amyloidogenic compounds of potential interest against Alzheimer’s and prion diseases. J Med Chem 55: 661-669.
- Muñoz-Torrero D, Pera M, Relat J, Ratia M, Galdeano C, et al. (2012) Expanding the multipotent profile of huprine-tacrine heterodimers as disease-modifying anti-Alzheimer agents. Neurodegener Dis 10: 96-99.
- Sola I, Viayna E, Gómez T, Galdeano C, Cassina M, et al. (2015) Multigram synthesis and in vivoefficacy studies of a novel multitarget anti-Alzheimer’s compound. Molecules 20: 4492-4515.
- Hu Y, Zhang J, Chandrashankra O, Ip FC, Ip NY (2013) Design, synthesis and evaluation of novel heterodimers of donepezil and huperzine fragments as acetylcholinesterase inhibitors. Bioorg Med Chem 21: 676-683.
- Heinrich M, Lee Teoh H (2004) Galanthamine from snowdrop--the development of a modern drug against Alzheimer's disease from local Caucasian knowledge. J Ethnopharmacol 92: 147-162.
- Marco-Contelles J, do Carmo Carreiras M, Rodríguez C, Villarroya M, García AG (2006) Synthesis and Pharmacology of Galantamine. Chem Rev 106: 116-133.
- Keglevich P, Szántay C, Hazai L (2016) The Chemistry of Galanthamine. Classical Synthetic Methods and Comprehensive Study on its Analogues. Mini Rev Med Chem 16: 1450-1461.
- Bores GM, Huger FP, Petko W, Mutlib AE, Camacho F, et al. (1996) Pharmacological evaluation of novel Alzheimer's disease therapeutics: acetylcholinesterase inhibitors related to galanthamine. J Pharmacol Exp Ther 277: 728-738.
- Sramek JJ, Frackiewicz EJ, Cutler NR (2000) Review of the acetylcholinesterase inhibitor galanthamine. Expert Opin Investig Drugs 9: 2393-2402.
- Atanasova M, Stavrakov G, Philipova I, Zheleva D, Yordanov N, et al. (2015) Galantamine derivatives with indole moiety: Docking, design, synthesis and acetylcholinesterase inhibitory activity. Bioorg Med Chem 23: 5382-5389.
- Sebestík J, Marques SM, Falé PL, Santos S, Arduíno DM, et al. (2011) Bifunctional phenolic-choline conjugates as anti-oxidants and acetylcholinesterase inhibitors. J Enzyme Inhib Med Chem 26: 485-497.
- Liu HR, Liu XJ, Fan HQ, Tang JJ, Gao XH, et al. (2014) Design, synthesis and pharmacological evaluation of chalcone derivatives as acetylcholinesterase inhibitors. Bioorg Med Chem 22: 6124-6133.
- Liu H, Fan H, Gao X, Huang X, Liu X, et al. (2016) Design, synthesis and preliminary structure-activity relationship investigation of nitrogen-containing chalcone derivatives as acetylcholinesterase and butyrylcholinesterase inhibitors: a further study based on Flavokawain B Mannich base derivatives. J Enzyme Inhib Med Chem 31: 580-589.
- Liu HR, Zhou C, Fan HQ, Tang JJ, Liu LB, et al. (2015) Novel potent and selective acetylcholinesterase inhibitors as potential drugs for the treatment of Alzheimer’s disease: Synthesis, pharmacological evaluation, and molecular modeling of amino-alkyl-substituted fluoro-chalcones derivatives. Chem Biol Drug Des 86: 517-522.
- de la Torre P, Saavedra LA, Caballero J, Quiroga J, Alzate-Morales JH, et al. (2012) A novel class of selective acetylcholinesterase inhibitors: synthesis and evaluation of (E)-2-(benzo[d]thiazol-2-yl)-3-heteroarylacrylonitriles. Molecules 17: 12072-12085.
- Ignasik M, Bajda M, Guzior N, Prinz M, Holzgrabe U, et al. (2012) Design, synthesis and evaluation of novel 2-(aminoalkyl)-isoindoline-1,3-dione derivatives as dual-binding site acetylcholinesterase inhibitors. Arch Pharm (Weinheim) 345: 509-516.
- Alpan AS, Parlar S, Carlino L, Tarikogullari AH, Alptüzün V, et al. (2013) Synthesis, biological activity and molecular modeling studies on 1H-benzimidazole derivatives as acetylcholinesterase inhibitors. Bioorg Med Chem 21: 4928-4937.
- Lu C, Guo Y, Yan J, Luo Z, Luo HB, et al. (2013) Design, synthesis, and evaluation of multitarget-directed resveratrol derivatives for the treatment of Alzheimer’s disease. J Med Chem 56: 5843-5859.
- Shi DH, Huang W, Li C, Wang LT, Wang SF (2013) Synthesis, biological evaluation and molecular modeling of aloe-emodin derivatives as new acetylcholinesterase inhibitors. Bioorg Med Chem 21: 1064-1073.
- Bukhari SN, Jantan I, Masand VH, Mahajan DT, Sher M, et al. (2014) Synthesis of α, β-unsaturated carbonyl based compounds as acetylcholinesterase and butyrylcholinesterase inhibitors: characterization, molecular modeling, QSAR studies and effect against amyloid β-induced cytotoxicity. Eur J Med Chem 83: 355-365.
- Canto RF, Barbosa FA, Nascimento V, de Oliveira AS, Brighente IM, et al. (2014) Design, synthesis and evaluation of seleno-dihydropyrimidinones as potential multi-targeted therapeutics for Alzheimer’s disease. Org Biomol Chem 12: 3470-3477.
- Parveen M, Malla AM, Alam M, Ahmad M, Rafiq S (2014) Stereoselective synthesis of Z-acrylonitrile derivatives: catalytic and acetylcholinesterase inhibition studies. New J Chem 38: 1655-1667.
- Friggeri L, De Vita D, Pandolfi F, Tortorella S, Costi R, et al. (2015) Design, synthesis and evaluation of 3,4-dihydroxybenzoic acid derivatives as antioxidants, bio-metal chelating agents and acetylcholinesterase inhibitors. J Enzyme Inhib Med Chem 30: 166-172.
- Tanarro CM, Gutschow M (2011) Hyperbolic mixed-type inhibition of acetylcholinesterase by tetracyclic thienopyrimidines. J Enzyme Inhib Med Chem 26: 350-358.
- Catto M, Berezin AA, Lo Re D, Loizou G, Demetriades M, et al. (2012) Design, synthesis and biological evaluation of benzo[e][1,2,4]triazin-7(1H)-one and [1,2,4]-triazino[5,6,1-jk]carbazol-6-one derivatives as dual inhibitors of beta-amyloid aggregation and acetyl/butyryl cholinesterase. Eur J Med Chem 58: 84-97.
- Liu S-J, Cui L-B, Xu H-L, Wang T-Y, Li S, et al. (2013) Design, synthesis, and biological evaluation of 7H-Thiazolo[3,2-b]-1,2,4-triazin-7-one derivatives as dual binding site acetylcholinesterase Inhibitors. Heterocycles 87: 2607-
- Liu S, Shang R, Shi L, Wan DC, Lin H (2014) Synthesis and biological evaluation of 7H-thiazolo[3,2-b]-1,2,4-triazin-7-one derivatives as dual binding site acetylcholinesterase inhibitors. Eur J Med Chem 81: 237-244.
- Liu S, Shang R, Shi L, Zhou R, He J, et al. (2014) Design, synthesis, and evaluation of 7H-thiazolo-[3,2-b]-1,2,4-triazin-7-one derivatives as dual binding site acetylcholinesterase inhibitors. Chem Biol Drug Des 84: 169-174.
- He Y, Yao PF, Chen SB, Huang ZH, Huang SL, et al. (2013) Synthesis and evaluation of 7,8-dehydrorutaecarpine derivatives as potential multifunctional agents for the treatment of Alzheimer’s disease. Eur J Med Chem 63: 299-312.
- Li Z, Wang B, Hou JQ, Huang SL, Ou TM, et al. (2013) 2-(2-indolyl-)-4(3H)-quinazolines derivates as new inhibitors of AChE: design, synthesis, biological evaluation and molecular modelling. J Enzyme Inhib Med Chem 28: 583-592.
- Darras FH, Wehle S, Huang G, Sotriffer CA, Decker M (2014) Amine substitution of quinazolinones leads to selective nanomolar AChE inhibitors with ‘inverted’ binding mode. Bioorg Med Chem 22: 4867-4881.
- Xing W, Fu Y, Shi Z, Lu D, Zhang H, et al. (2013) Discovery of novel 2,6-disubstituted pyridazinone derivatives as acetylcholinesterase inhibitors. Eur J Med Chem 63: 95-103.
- Saleem M, Rafiq M, Hanif M, Rama NH, Seo S-Y, et al. (2012) Synthesis, Urease and Acetylcholine Esterase Inhibition Activities of Some 1,4-Disubstituted Thiosemicarbazides and their 2,5-Disubstituted Thiadiazoles. Korean Chem Soc 33: 2741-2747.
- Skrzypek A, Matysiak J, Karpi?ska MM, Niewiadomy A (2013) Synthesis and anticholinesterase activities of novel 1,3,4-thiadiazole based compounds. J Enzyme Inhib Med Chem 28: 816-823.
- Skrzypek A, Matysiak J, Niewiadomy A, Bajda M, Szyma?ski P (2013) Synthesis and biological evaluation of 1,3,4-thiadiazole analogues as novel AChE and BuChE inhibitors. Eur J Med Chem 62: 311-319.
- Liu W-w, Li Q-x, Shi D-h, Cao Z-l, Cheng F-c, et al. (2015) Synthesis, characterization, and biological evaluation of some novel glycosyl 1,3,4-thiadiazole derivatives as acetylcholinesterase inhibitors. Heterocycles 91: 275-286.
- Ismail MM, Kamel MM, Mohamed LW, Faggal SI, Galal MA (2012) Synthesis and biological evaluation of thiophene derivatives as acetylcholinesterase inhibitors. Molecules 17: 7217-7231.
- Basiri A, Murugaiyah V, Osman H, Kumar RS, Kia Y, et al. (2013) An expedient, ionic liquid mediated multi-component sy
Citation: Saxena AK, Saini R (2018) The Structural Hybrids of Acetylcholinesterase Inhibitors in the Treatment of Alzheimer's Disease: A Review. J Alzheimers Neurodegener Dis 4: 015.
Copyright: © 2018 Ratni Saini, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.