Journal of Stem Cells Research Development & Therapy Category: Medical
Type: Research Article
Tissue Complex of Adult Pancreatic Duct and Vascular Endothelial Cells Promotes In Vitro Differentiation into Insulin-Producing Cells
*Corresponding Author(s):
Jun KanamuneDepartment Of Organ Reconstruction, Field Of Clinical Application, Institute For Frontier Medical Sciences, Kyoto University, Japan
Tel:+81 757514866,
Email:jkanamune@frontier.kyoto-u.ac.jp
Received Date: Mar 31, 2015
Accepted Date: Jun 03, 2015
Published Date: Jun 17, 2015
Abstract
In adult pancreas, a number of tissue-residing stem-like cells have been isolated, among which epithelial duct cells are known to have a certain degree of plasticity to become insulin-producing cells. Since the epithelial duct cells during pancreatic development have a close contact with endothelial cells, we asked if adult pancreatic duct cells cultured in close contact with vascular endothelial cells will be enhanced of endocrinal differentiation. We isolated duct-enriched fractions of cells from enzyme-digested pancreatic debris tissues devoid of islets and expanded them in serum-free media. When these duct cells upon differentiation were directly mixed with vascular endothelial cells separately cultured from the pancreatic debris tissues, the resulting spherical tissues contained an increased proportion of insulin-positive cells. When transplanted into streptozotocin induced diabetic mice, the spherical tissues normalized the blood glucose levels in about 10% of the recipients and allowed the rest of the recipients to survive for a period of 50 days until they were sacrificed. To characterize purified duct cells in their potential for In Vitroproliferation and differentiation, we sorted cells in the pancreatic debris tissues using Aldefluor dye. The Aldefluor-E-cadherin double positive cells proliferated with limited magnitudes, but the proliferation was significantly enhanced to generate monolayers when co-cultured with the stellate cells separately isolated from the same pancreatic debris tissues. These epithelial cells further In Vitro differentiated into mostly glucagon-positive cells and small fractions of insulin-positive cells. Therefore, the ex vivo cultures of both duct and stellate cells proliferate together, and upon differentiation as a tissue complex with vascular endothelial cells, they generate spherical tissues containing pancreatic endocrine cells with a potential to cure diabetes.
INTRODUCTION
Stem-like cells isolated from adult pancreatic cells are known to exhibit a certain degree of plasticity that under In Vitro conditions they potentially differentiate into pancreatic endocrine cells including β cells [1-3]. Despite the therapeutic potential of these tissue-residing stem-like cells, it has not yet been implemented as a proof of concept for transplantation-based cell therapy for diabetes based on two major reasons. First, culturing methods have not yet been optimized in collecting, expanding and differentiating sufficient amounts of these cells that can normalize glucose levels in the diabetic subjects. To optimize the In vitro differentiation process, we examined the role of vascular endothelial cells in a co-culture environment. Second, the identity of such tissue-residing stem-like cells has not yet been clarified to ensure the reproducibility of the optimization. We used Aldefluor dye to isolate and culture a group of pancreatic ductal cells, which have been reported to differentiate into pancreatic endocrine cells [4].
Vascular endothelial cells are known to be involved in pancreatic organogenesis. An important seminal work indicated a direct role of vascular endothelial cells in expressing Pdx1, a master transcription factor to engage in the expansion of endocrine precursor population [5]. In organogenesis, contacts with endothelial cells allow endoderm to generate a pancreatic rudiment tissue expressing Pdx1 [6-10]. Moreover, biology of native islets indicates the importance of endothelial cells, as a paracrine source that secrete extracellular matrices [11] as well as soluble factors that promote islet survival and function [12,13].
Based on the view that adult pancreatic stem-like cells partially recapitulate the organogenesis in the course of differentiation into endocrinal cells [14,15], vascular endothelial cells possibly enhance their differentiation in the In vitro regeneration model [16]. Using a pancreatic injury and regeneration model in adult mice, we previously found that doxorubicin-induced perturbation of vascular endothelial tissues significantly depleted of a population of CD133-positive pancreatic stem cells [17], indicating that vascular endothelial cells are involved in pancreatic regeneration.
In the light of these background data, we hypothesized that a direct contact between vascular endothelial cells and pancreatic stem duct cells would enhance the differentiation of the latter into endocrinal cells. In support of the hypothesis, the tissue complex of these two different cell populations that were isolated and expanded from a common tissue source of the pancreas showed increased proportions of insulin-producing cells and insulin-secreting abilities. The tissue complex when transplanted into the kidney normalized glucose levels in some individuals of diabetic mice.
In the course of this study, we investigated the role of another cell of mesodermal origin, stellate cell, in the In vitro proliferation of the pancreatic stem-like cells. Stellate cells and their role as a promoter of cell proliferation are well recognized particularly in the pathological setting of pancreatic inflammation. Stellate cells upon experimental pancreatic injuries are known to proliferate in the tissue regeneration [18]. Because stellate cells produce mitogens such as cytokines and growth factors involved in precursory conditions leading to oncogenesis [19], they are thought to function as a paracrine source of similar mitogens in regenerating pancreas.
We tested if stellate cells would promote the murine adult pancreatic stem-like cells to proliferate in a co-culture model. The stellate cells kept in a transwell insert substantially increased the proliferation of an Aldeflour-positive pancreatic stem cells cultured in a common medium, which eventually established a monolayer of epithelial cell population with a potency to differentiate into endocrinal cells.
Vascular endothelial cells are known to be involved in pancreatic organogenesis. An important seminal work indicated a direct role of vascular endothelial cells in expressing Pdx1, a master transcription factor to engage in the expansion of endocrine precursor population [5]. In organogenesis, contacts with endothelial cells allow endoderm to generate a pancreatic rudiment tissue expressing Pdx1 [6-10]. Moreover, biology of native islets indicates the importance of endothelial cells, as a paracrine source that secrete extracellular matrices [11] as well as soluble factors that promote islet survival and function [12,13].
Based on the view that adult pancreatic stem-like cells partially recapitulate the organogenesis in the course of differentiation into endocrinal cells [14,15], vascular endothelial cells possibly enhance their differentiation in the In vitro regeneration model [16]. Using a pancreatic injury and regeneration model in adult mice, we previously found that doxorubicin-induced perturbation of vascular endothelial tissues significantly depleted of a population of CD133-positive pancreatic stem cells [17], indicating that vascular endothelial cells are involved in pancreatic regeneration.
In the light of these background data, we hypothesized that a direct contact between vascular endothelial cells and pancreatic stem duct cells would enhance the differentiation of the latter into endocrinal cells. In support of the hypothesis, the tissue complex of these two different cell populations that were isolated and expanded from a common tissue source of the pancreas showed increased proportions of insulin-producing cells and insulin-secreting abilities. The tissue complex when transplanted into the kidney normalized glucose levels in some individuals of diabetic mice.
In the course of this study, we investigated the role of another cell of mesodermal origin, stellate cell, in the In vitro proliferation of the pancreatic stem-like cells. Stellate cells and their role as a promoter of cell proliferation are well recognized particularly in the pathological setting of pancreatic inflammation. Stellate cells upon experimental pancreatic injuries are known to proliferate in the tissue regeneration [18]. Because stellate cells produce mitogens such as cytokines and growth factors involved in precursory conditions leading to oncogenesis [19], they are thought to function as a paracrine source of similar mitogens in regenerating pancreas.
We tested if stellate cells would promote the murine adult pancreatic stem-like cells to proliferate in a co-culture model. The stellate cells kept in a transwell insert substantially increased the proliferation of an Aldeflour-positive pancreatic stem cells cultured in a common medium, which eventually established a monolayer of epithelial cell population with a potency to differentiate into endocrinal cells.
MATERIALS AND METHODS
Pancreatic tissue collection
Murine pancreata were harvested after intra-ductally injecting 2.0 to 2.5ml 0.1% Collagenase S-1 (Nitta Gelatin, Inc.) in HBSS (Sigma-Aldrich Co.) into the pancreas of 8 to 14 weeks old mice. Groups of three harvested pancreata were incubated in 5ml HBSS at 37°C for 20min, cooled on ice, shaken vigorously for 20 seconds with additional 10ml RPMI 1640 (Invitrogen) and passed through a 1.0mm steel mesh to remove large clumps of debris. The pancreatic digests were thrice washed with 50ml RPMI 1640 by centrifuging at 250xg at 4°C for 2min. About 0.5ml of the tissue pellet was re-suspended with a 15ml histopaque 1077 (Sigma-Aldrich) in a 50ml conical tube, and the suspension was gently overlaid with 15ml of the mixture and further with 5ml of RPMI 1640, followed by a centrifugation at 400xg with slow start and stop at 4°C for 20min. A hazy layer in the middle density gap containing mostly islets was removed, and left-over tissues, “pancreatic debris,” were twice washed with 50ml RPMI 1640 by centrifuging at 250xg at 4°C for 2min.
Non-endocrinal epithelial cell culture
The pancreatic debris from each pancreas was seeded with a serum-containing medium, RPMI 1640 supplemented with 10% heat-inactivated FCS (BioWest, Inc.) and PenStrep (final concentration=50/50, Wako Chemicals Co.) in each of 10cm dishes (Greiners, Inc.) pre-coated with 0.2mg/ml rat tail collagen I (BD Biosciences) and cultured at 37°C and 5% CO2 for 24 to 36hrs. After this initial incubation, floating tissue debris was removed, and the medium was replaced with a serum-free medium, DMEM/Ham’s F-12 containing 7.8mM D-glucose (Wako Chemicals Co.), 1g/l ITS (final concentrations: 5μg/l insulin, 5μg/l transferrin, 5ng/l sodium selenite) (MP Biomedicals), 0.2% BSA (Sigma-Aldrich Co.), 10mM nicotinamide (Sigma-Aldrich Co.), 10ng/ml recombinant human KGF (Wako Chemicals Co.) and PenStrep (50/50, Wako Chemicals Co.), and cultured for three days.
About 60% of the media was subsequently replenished in every 2-3 days. With this serum-free medium culture, scattered colonies of Non-Endocrinal Epithelial Cells (NEECs) were passaged into separate culture dish, so that they eventually formed dense cell clusters. After this stage, the cultures were occasionally treated with diluted dithizone solution to stain any remaining islet tissue contaminants to be manually removed. For passaging, 50 to 70%-confluent NEECs were washed twice with PBS and removed by incubating with Accutase (Innovative Cell Technologies, Inc.) at RT for less than 3min. The cells were gently removed by adding the culture medium, passed through a 40μm mesh to remove large tissue clumps, and pelleted cells were re-suspended with the same serum-free medium and seeded at or above 5.0 x 106 cells/ml on a collagen I-coated 10cm dish or a small fraction of these on 4-well slide chambers (Biocoat Cellware, BD Labware) for immunohistochemical analyses.
About 60% of the media was subsequently replenished in every 2-3 days. With this serum-free medium culture, scattered colonies of Non-Endocrinal Epithelial Cells (NEECs) were passaged into separate culture dish, so that they eventually formed dense cell clusters. After this stage, the cultures were occasionally treated with diluted dithizone solution to stain any remaining islet tissue contaminants to be manually removed. For passaging, 50 to 70%-confluent NEECs were washed twice with PBS and removed by incubating with Accutase (Innovative Cell Technologies, Inc.) at RT for less than 3min. The cells were gently removed by adding the culture medium, passed through a 40μm mesh to remove large tissue clumps, and pelleted cells were re-suspended with the same serum-free medium and seeded at or above 5.0 x 106 cells/ml on a collagen I-coated 10cm dish or a small fraction of these on 4-well slide chambers (Biocoat Cellware, BD Labware) for immunohistochemical analyses.
Vascular endothelial cell culture
The floating tissue debris removed from the culture of pancreatic discards was seeded with EBM medium (Lonza, Inc.) in a 10cm dish coated with 0.1mg/ml rat tail collagen I. The adherent cells on the dish were cultured for 10 days and removed by Accutase at RT for 5-10min. Washed and suspended cells were purified by streptavidin-conjugated microbeads (Miltenyi Biotec, Inc.). Magnetically captured cells eluting from a separation column constituting mostly Vascular Endothelial Cells (VECs) were cultured with the same medium in collagen I-coated T25 flasks (Corning, Inc.) for further expansion. Small fractions of these cells were stained with PE-conjugated anti-mouse CD31 antibody (eBiosciences) and verified of their purity to be over 90%.
Formation of tissue complex
NEECs and VECs were removed from culture containers by Accutase treatments as described earlier and mixed at the cell number ratio of approximately 7:3, respectively. The cell mixture was suspended in the ultralow attachment 6-well plate (Greiner, Inc.) with a differentiation medium containing DMEM/Ham’s F-12, 20mM D-glucose, 2% (v/v) FCS, 2ng/ml Activin A (PeproTech, Inc.), 5ng/ml Betacellulin (PeproTech, Inc.), 20ng/ml HGF (PeproTech, Inc.), 20ng/ml IGF-1 (R&D Systems, Inc.), 5ng/ml KGF (Wako Chemicals Co.), 20ng/ml Exendin-4 (Sigma-Aldrich Co.), 20ng/ml VEGF (PeproTech, Inc.), 10mM nicotinamide, 0.1mM ascorbic acid and 1mM CaCl2. The suspended cell mixture was then subjected to a slow swirling motion (20rpm) at 37°C and 5% CO2 for 18 to 24hrs until spherical tissue clusters, tissue complexes, formed, which were then incubated with the medium without CaCl2 in the culture container horizontally placed without motion. The culture medium was replenished every 1-2 days for the following 7 days, and switched to that without Activin A, Betacellulin and KGF for additional 14 days. After the differentiation culture period, tissue complexes were measured of diameters and calculated of Islet Equivalents (IEQs); one IEQ equaled to a tissue complex of 200μm in diameter. For tissues smaller or larger than this, IEQs were estimated based on their volume.
Immunohistochemistry
The spherical tissue complexes were fixed in 4% PFA in phosphate buffer at 4°C for 18hrs, washed in PBS at 4°C for 24hrs, dehydrated in alcohol series and xylene and embedded in paraffin. Four μm paraffin sections were de-waxed on glass slides, rehydrated in alcohol series and PBS and permeabilized with 0.1% Triton X-100. The tissue sections were blocked with the blocking solution at RT for 1hour, and treated with guinea pig anti-insulin antibody (Dako) (1:400) at RT for 1 hour. The tissue sections were then washed and treated with Cy3-conjugated donkey anti-guinea pig IgG secondary antibody (Chemicon) (1:1000) in the blocking solution. To identify stellate cells cultured from pancreatic discards were treated with anti-α-SMA antibody (Dako) (1:100) and Alexa Fluor 488-conjugated donkey anti-mouse antibody (Life Technologies) (1:500). Aldefluor-E-cadherin double positive pancreatic cells co-cultured with the stellate cells were identified by FITC-conjugated anti-mouse E-cadherin antibody (BD Pharmingen) (1:400). After incubated in differentiation media, insulin- and glucagon-positive cells were labeled with the anti-insulin antibody and rabbit anti-glucagon antibody (Dako) (1:400) followed by Alexa Fluor-conjugated donkey anti-rabbit IgG secondary antibody (Chemicon) (1:1000), respectively. Cells on slides were counter-stained with DAPI, mounted and observed with a fluorescent confocal microscope. The proportions of positively-stained cells were determined with 7 fields for each slide sample. To evaluate the epithelial cells’ potential toward endocrinal differentiation, the cells in slide chambers were incubated with cyclopamine KAAD (Cycl-K) (Calbiochem) and All-trans Retinoic Acid (RA) (Wako Chemicals) or left untreated. These cells were then fixed with 4% paraformaldehyde, washed with PBS and antigens were uncovered by trypsin before blocking and treatment with anti-Ngn3 rabbit IgG (Millipore) (400:1) and Alexa Fluor 488-conjugated anti-rabbit goat IgG secondary antibody (Chemicon) (800:1). Nuclei were counter stained with DAPI to confirm the nuclear localization of Ngn3.
RNA isolation and semi-quantitative RT-PCR
Total RNA was extracted from about 1.0x106 NEECs and tissue complexes by RNeasy Mini Kit (Qiagen), and DNA contaminants were removed by RNase-free DNase I Set (Qiagen). The RNA (50ng) was reverse transcribed using Super Script III (Invitrogen, Inc.) with random primers (Invitrogen), and synthesized cDNAs were treated with RNase H (Wako Chemicals) to remove any RNA contaminants. PCR was performed with 50ng cDNA with temperature cycling conditions: one cycle of 95°C for 2min, followed by 28-40 cycles of 95°C for 30sec, 45-59°C for 30sec, and 72°C for 45sec. The number of cycles and annealing temperatures were adjusted for detecting the amount of amplicons that increased linearly. Primer pairs used to detect murine genes are listed in table 1. The relative amounts of amplified PCR products were analyzed in comparison with those of the internal control, GAPDH, by gel-electrophoresis with 2% agarose gel and 0.001% ethidium bromide.
| Genes | GenBank no. | Forward primer | Reverse primer | Size |
| Glucagon | NM_008100.3 | CCACTCACAGGGCACATTCA | GTCCCTGGTGGCAAGATTGT | 343 |
| Hes1 | NM_008235.2 | TCAACACGACACCGCACAAACC | GGTACTTCCCCAACACGCTCG | 270 |
| HNF6 | NM_008262.3 | GCAATGGAAGTAATTCAGGGCAG | CATGAAGAAGTTGCTGACAGTGC | 461 |
| Ins1 | NM_008386.3 | GCTGGTAGAGGGAGCAGATG | CAGAGACCATCAGCAAGCAG | 341 |
| Ins2 | NM_001185084.1 | CCCTGCTGGCCCTGCTCTT | AGGTCTGAAGGTCACCTGCT | 213 |
| Neuro D | NM_010894.2 | GTCCCAGCCCACTACCAATT | CGGCACCGGAAGAGAAGATT | 440 |
| Ngn3 | NM_009719.6 | TCCTCGGAGCTTTTCTACGA | TCTGAGTCAGTGCCCAGATG | 598 |
| Nkx6.1 | NM_144955.2 | CCGGTCGGACGCCCATC | GAGGCTGCCACCGCTCGATTT | 467 |
| Notch1 | NM_008714.3 | TTACAGCCACCATCACAGCCA | AATGCCCTGGACCAATCAGA | 380 |
| Pdx1 | NM_008814.3 | CCACCCCAGTTTACAAGCTC | TGTAGGCAGTACGGGTCCTC | 325 |
| Ptf1a | NM_018809.2 | AACCAGGCCCAGAAGGTTAT | CCTCTGGGGTCCACACTTTA | 169 |
| Sox9 | NM_011448.4 | GGCGGAGGAAGTCGGTGAAG | GGGTGCGGTGCTGCTGAT | 449 |
| GAPDH | NM_008084.2 | AACTTTGGCATTGTGGAAGG | ACACATTGGGGGTAGGAACA | 223 |
Evaluation of insulin-secreting function
To evaluate insulin-secreting function of the generated tissue complexes, 20 IEQs of the tissues after 20 days of maturation were pre-incubated with 200μl KREBS ringer solution supplemented with 0.1% BSA, 10mM Hepes, PenStrep and 5mM D-glucose, a low glucose solution, in a well of a U-shape bottom 96-well plate (Nalgene Nunc International) at 37°C and 5% CO2 for 1hour. Twenty IEQs of native islets were also pre-incubated but for overnight (18hr) before glucose stimulation to acclimate them to the experimental conditions. Tissue complexes or native islets were carefully washed 2-3 times and incubated with the same amount of fresh solution at 37°C and 5% CO2 for 2hr. After incubation, 150μl of the low glucose solution was collected from the wells, and the tissues were similarly washed 2-3 times and incubated with the same amount of fresh ringer solution but with 20mM D-glucose, a high glucose solution, followed by collecting 150μm of the solution. The KREBS ringer solution samples were stored at -30°C. These were later thawed, centrifuged briefly to take 50μl from each sample and subjected to mouse C-peptide assay by ELISA (Shibayagi Co.) according to the manufacturer’s instructions.
In vivo functional evaluation of tissue complex
Overnight-fasted mice were rendered diabetic by intraperitoneally injecting streptozotocin (160mg/kg body weight), and blood glucose levels were measured. Mice with the blood glucose levels of above 400mg/dl on two consecutive days were considered as diabetic. The ventro-lateral peritoneum of diabetic mice under anesthesia was surgically opened and transplanted with 700 to 1500 IEQs of the In vitro-generated islet-like spherical tissues under a kidney capsule with a small catheter. The other kidney was left intact. The incision was closed by a double-closure technique before the mice were released from anesthesia. Operated mice were followed up by measuring body weights and blood glucose levels by Accu Chek Active (Roche Diagnostics) for 50 days.
Sorting of Aldefluor-positive cells by FACS
The pancreatic tissue debris was washed twice with PBS and further digested with Accutase (Stem Cell Technologies, Inc.) into single cells at RT for 30 min. From this cell population, cells with high aldehyde dehydrogenase expression were labeled by using the Aldefluor Kit (Stem Cell Technologies) after the protocol by [4]. Briefly, the single cells were incubated in Aldefluor buffer for 40min at 37°C. A negative control was made by arresting the enzymatic activity with diethylaminobenzaldehyde. The enzyme-treated cells were then incubated with APC-conjugated anti-E-cadherin antibody (BioLegend) (1:400) for 30min. The sorting gate for the Aldefluor-positive cells was determined above the lower threshold set by the green fluorescent emission of the negative control. Flow cytometry was performed using a FACS Aria II (Becton Dickinson) flow cytometer.
Promotion of Aldefluor-positive stem-like cells by co-culture with stellate cells
Sorted Aldefluor-positive cells were cultured in 12-well plates (500cells/well) coated with 0.2mg/ml rat collagen I (BD Pharmingen, Inc.) containing DMEM/Ham’s F12 (7.8mM D-glucose) with 0.025% ITS (MP Biomedicals, Inc.), 1.88mg/ml of human insulin and transferrin (Wako Chemicals, Inc.), 10ng/ml recombinant human HGF (Wako Chemicals, Inc.), 10ng/ml recombinant human EGF (Invitrogen Gibco, Inc.), Penicillin + Gentamycin (50/50) and 1.0µM ROCK inhibitor (Y-27632, Calbiochem, Inc.). To enhance their proliferation, these cells were co-cultured in 12 well plates with pancreatic stellate cells separately isolated from murine pancreatic mesenchyme and seeded into a transwell insert coated with Collagen I (1µm pore size, Millipore, Inc.). These stem-like cells were passaged in this co-culture environment.
RESULTS
In vitro cultures of non-endocrinal epithelial cells
Colonies of epithelial cells emerged from scattered cells and tissue clumps attaching on the collagen-coated dish surface (Figure 1A). These cells measuring 30-50μm in diameter proliferated on first 3-5 days after the serum-containing culture media were switched to serum-free media (Figure 1B). They subsequently formed a few colonies of dense monolayer clusters of cells each measuring 20-40μm in diameter with sustained general epithelial morphology for 7 days of culture (Figure 1C). These epithelial cell clusters were surrounded by sparsely-distributed tissue clumps, which were eliminated by a nylon mesh when the epithelial cells were passaged. The epithelial cell populations showed the mRNA expression of Sox9 (Figure 2), signifying ductal and/or centroacinar lineages maintained over several passages. When stimulated with all-trans retinoic acid and cyclopamine KAAD for 24 hours, nuclear Ngn3 expression increased (Figure 3), indicating that these cells have a potential to differentiate into endocrine cells. Gene expression profiles of the epithelial cells reflect an early stage of differentiation into pancreatic endocrinal cells (Figure 4).
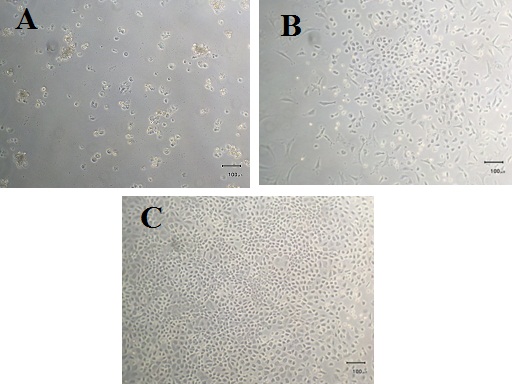 Figure 1: Cells and tissue fragments attaching to the dish surface after the culture in a serum-containing medium for 36 hrs (A). Cell colonies were induced to grow in a serum-free medium (B) to form a dense monolayer of cells of an epithelial morphology within 7 days of culture in the same serum-free medium (C).
Figure 1: Cells and tissue fragments attaching to the dish surface after the culture in a serum-containing medium for 36 hrs (A). Cell colonies were induced to grow in a serum-free medium (B) to form a dense monolayer of cells of an epithelial morphology within 7 days of culture in the same serum-free medium (C).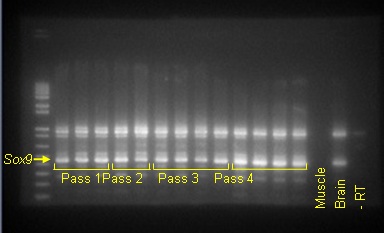 Figure 2: Expressions of Sox9 mRNA in the populations of cells passaged in time sequence. Each lane indicates the signal of a different cell population. A negative (Muscle tissue), positive cDNA controls (Brain tissue) and a PCR product without Reverse Transcriptase (-RT) are shown on the right.
Figure 2: Expressions of Sox9 mRNA in the populations of cells passaged in time sequence. Each lane indicates the signal of a different cell population. A negative (Muscle tissue), positive cDNA controls (Brain tissue) and a PCR product without Reverse Transcriptase (-RT) are shown on the right.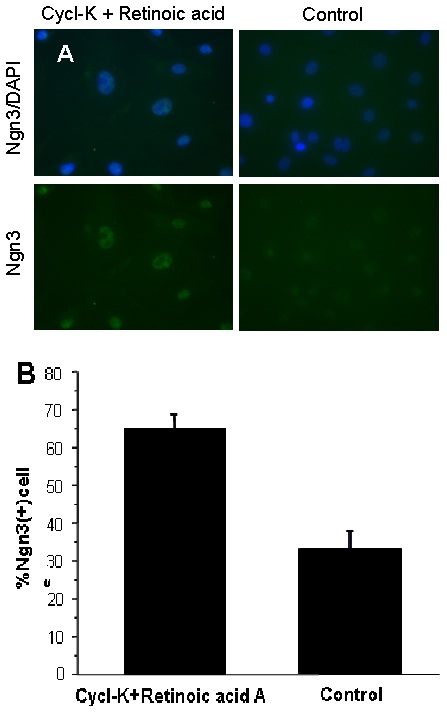 Figure 3: Nuclei of NEECs show the expression of Ngn3 for those cultured with all-trans retinoic acid A and Cyclopamine KAAD for 24 hrs or left untreated (A). Nuclei were counter-stained with DAPI. These cells show a significantly greater proportion of Ngn3-positive cells than those untreated (B) (Unpaired Student-t test, n=3 cell samples per treatment, p<0.0001).
Figure 3: Nuclei of NEECs show the expression of Ngn3 for those cultured with all-trans retinoic acid A and Cyclopamine KAAD for 24 hrs or left untreated (A). Nuclei were counter-stained with DAPI. These cells show a significantly greater proportion of Ngn3-positive cells than those untreated (B) (Unpaired Student-t test, n=3 cell samples per treatment, p<0.0001).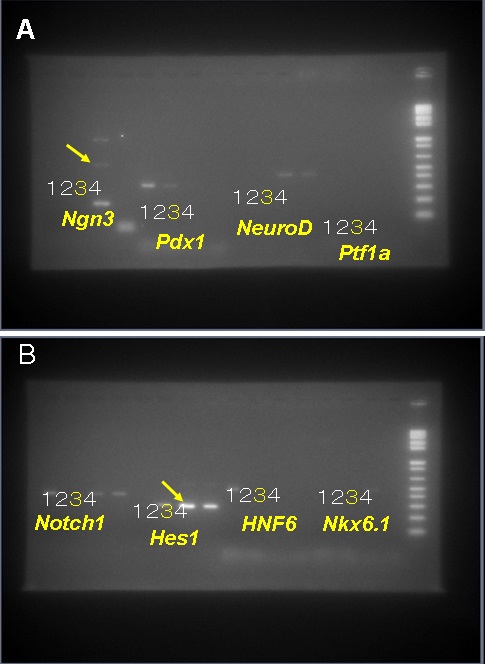 Figure 4: Relative expressions of genes: Ngn3, Pdx1, NeuroD and Ptf1a (A) and Notch1, Hes1, HNF6 and Nkx6.1 (B) based on semi-quantitative RT-PCR of the NEECs. The numbers 1-4 indicate the tissues or cell cultural samples used for the analyses: 1, pancreatic discards; 2, NEECs after 5 days of culture in serum-free media; 3, NEECs cultured with media containing all-trans retinoic acid A and cyclopamine KAAD for 24 hrs; and 4, NEECs’ culture control of 3. Arrows highlight that Ngn3 and Hes1 expressions seem synchronized in heterogeneous cell populations in the cultures.
Figure 4: Relative expressions of genes: Ngn3, Pdx1, NeuroD and Ptf1a (A) and Notch1, Hes1, HNF6 and Nkx6.1 (B) based on semi-quantitative RT-PCR of the NEECs. The numbers 1-4 indicate the tissues or cell cultural samples used for the analyses: 1, pancreatic discards; 2, NEECs after 5 days of culture in serum-free media; 3, NEECs cultured with media containing all-trans retinoic acid A and cyclopamine KAAD for 24 hrs; and 4, NEECs’ culture control of 3. Arrows highlight that Ngn3 and Hes1 expressions seem synchronized in heterogeneous cell populations in the cultures.Generation of tissue complex from NEECs and VECs
The swirling suspension culture of NEECs and VECs readily generated the tissue complex starting with small clusters of a few cells, which then attached together to form larger spherical tissues of 100-250μm in diameter (Figure 5A-C). Spherical tissues of similar size groups were also generated from NEECs alone. During initial 7 days after the start of differentiation culture, the surface texture of the spheres with or without VECs became smoother, and their cores turned brownish red in color. As these tissues were further cultured in the differentiation medium, small transparent tissue parts budded out from the spheres around 15 days of culture, and tissues often merged to form larger tissue clumps (Figure 5D). Approximately 700 to 1000 IEQs of the spherical tissues were generated from the tissue complex derived from 2-3 pancreata. These were greater in amounts than those generated solely from the NEECs (Figure 5E), which were insufficient for transplantation experiments.
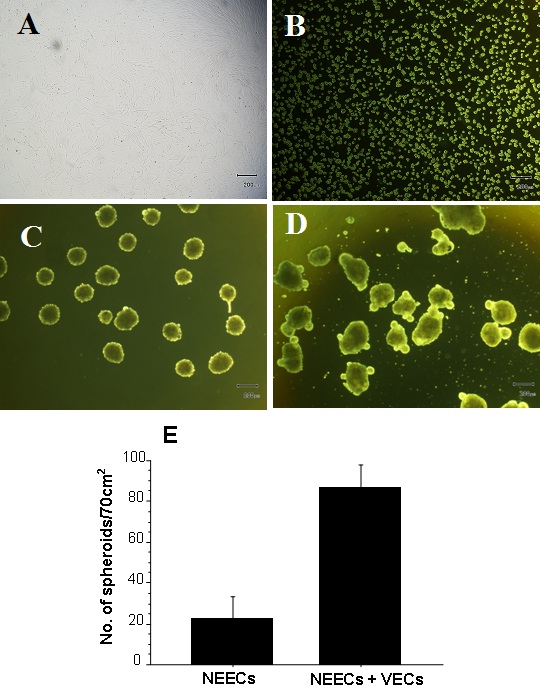 Figure 5: Generation of spherical tissues from the tissue complex of NEECs and VECs. VECs purified by magnetic beads using anti-CD31 antibody and expanded (A); Mixed cell suspension consisting of 80% NEECs and 20% VEC in a differentiation medium prior to incubation (B); spherical tissues after incubation with a swirling motion for 24 hrs (C); and those in maturation: smaller tissue parts budded out, while some spherical tissues merged to form larger tissue clumps (D). Size bars represent 200μm. Numbers of spherical tissues (200-400μm diameter) generated either from NEECs alone or a tissue complex of NEECs and VECs (E) (Unpaired t-test, n=3 groups of incubated cells, p<0.0001).
Figure 5: Generation of spherical tissues from the tissue complex of NEECs and VECs. VECs purified by magnetic beads using anti-CD31 antibody and expanded (A); Mixed cell suspension consisting of 80% NEECs and 20% VEC in a differentiation medium prior to incubation (B); spherical tissues after incubation with a swirling motion for 24 hrs (C); and those in maturation: smaller tissue parts budded out, while some spherical tissues merged to form larger tissue clumps (D). Size bars represent 200μm. Numbers of spherical tissues (200-400μm diameter) generated either from NEECs alone or a tissue complex of NEECs and VECs (E) (Unpaired t-test, n=3 groups of incubated cells, p<0.0001).Tissues generated with and without VECs
Immunohistochemical analyses showed that the spherical tissues with VECs contained a greater proportion of insulin-positive cells than those without VECs (Figure 6A-C). The distribution of these insulin-positive cells were scattered throughout the spherical tissues unlike a central distribution usually observed in murine islets. The greater proportion of insulin-producing cells for the tissues with VECs was paralleled by the greater expression levels of Ins1, Ins2 and Glucagon than those without VECs (Figure 7).
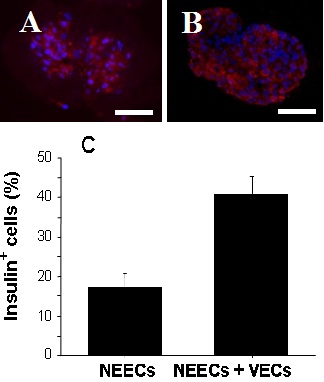 Figure 6: Spherical tissues matured for 20 days. Tissues generated from NEECs alone (A) and those generated from a tissue complex of NEECs and VECs (B). Size bars represent 30μm. Spherical tissues generated from the tissue complex contained more insulin-positive cells than those of NEECs alone (C) (n=5 representative spherical tissues for each group, Unpaired t-test, p<0.001).
Figure 6: Spherical tissues matured for 20 days. Tissues generated from NEECs alone (A) and those generated from a tissue complex of NEECs and VECs (B). Size bars represent 30μm. Spherical tissues generated from the tissue complex contained more insulin-positive cells than those of NEECs alone (C) (n=5 representative spherical tissues for each group, Unpaired t-test, p<0.001).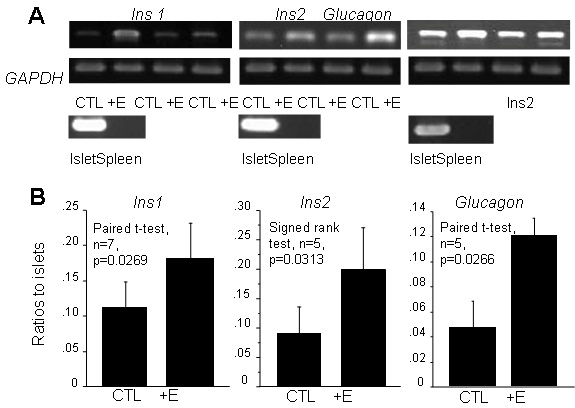 Figure 7: Ins1, Ins2 and Glucagon mRNA expressions amplified by semi-quantitative PCR. (A) Spherical tissues generated from a tissue complex of NEECs and VECs (+E) show greater relative expression intensities than those generated from NEECs alone (CTL); tissues from two different experiments are shown for each gene. Expressions of corresponding positive (Islet) and negative (Spleen) controls are shown below. (B) Expressions of these genes are greater for the tissue complex (+E) than for the tissues generated from NEECs alone (CTL) (For Ins1 and Glucagon, paired t-test was performed; for Ins2, data homogeneity did not warrant for parametric analyses; thus, singed rank test was performed).
Figure 7: Ins1, Ins2 and Glucagon mRNA expressions amplified by semi-quantitative PCR. (A) Spherical tissues generated from a tissue complex of NEECs and VECs (+E) show greater relative expression intensities than those generated from NEECs alone (CTL); tissues from two different experiments are shown for each gene. Expressions of corresponding positive (Islet) and negative (Spleen) controls are shown below. (B) Expressions of these genes are greater for the tissue complex (+E) than for the tissues generated from NEECs alone (CTL) (For Ins1 and Glucagon, paired t-test was performed; for Ins2, data homogeneity did not warrant for parametric analyses; thus, singed rank test was performed).Glucose stimulation tests
Glucose stimulation tests indicated that the spherical tissues generated with VECs secreted C-peptide in average 5.62 and 8.56ng/μg DNA in low and high glucose levels, respectively (Figure 8). Although these values were 21% and 17% of the native islets (n=3 sample tissues, Low glucose: 26.7±15.4ng/μg DNA; High glucose: 51.1±18.6ng/μg DNA), respectively, they were significantly higher than those secreted by the spherical tissues generated without VECs. The tissues with VECs also showed greater glucose responsiveness than those without them (Figure 8).
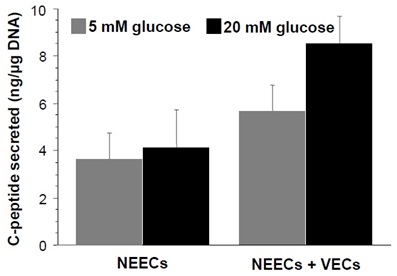
Figure 8: Spherical tissues secreted C-peptide upon stimulation with low (5mM) and high (20mM) glucose concentrations in KREBS buffer medium. Approximately 200 IEQs/sample were incubated in 250μl buffer. Spherical tissues generated from the tissue complex of NEECs and VECs secreted larger amounts of C-peptide with greater glucose responsiveness than those generated from NEECs alone (Two-way ANOVA: effect= NEECs+VECs vs. NEECs alone, F=32.5, n=5 groups, p<0.0001; effect= low glucose vs. high glucose concentrations, F=9.1, n=5 groups, p=0.0082).
In vivo glucose-controlling ability of the generated tissues
To test the glucose-controlling ability of the spherical tissues generated by a tissue complex of NEECs and VECs, the spherical tissues were transplanted to diabetic mice, whose blood glucose levels were monitored. Of the total of 9 diabetic mice transplanted, 5 mice exhibited gradually-lowered glucose levels for 4 weeks after the transplantation, among which one mouse reached normoglycemia (Figure 9A). The 4 other mice showed high glucose levels often above 600mg/dl but survived throughout the experimental period of 7 weeks before being sacrificed, while a control mouse without transplants died after 32 days following diabetic induction before the end of the experimental period (Figure 9B). Immunohistochemical analysis of the kidneys transplanted with the spherical tissues showed flattened clusters of insulin-positive cells in the narrow gaps between the kidney mesenchyme and a kidney capsule membrane (Figure 10). For the spherical tissues generated from NEECs alone, the tissue amounts often did not reach the minimum 700 IEQ’s, and were not used for transplantation experiments.
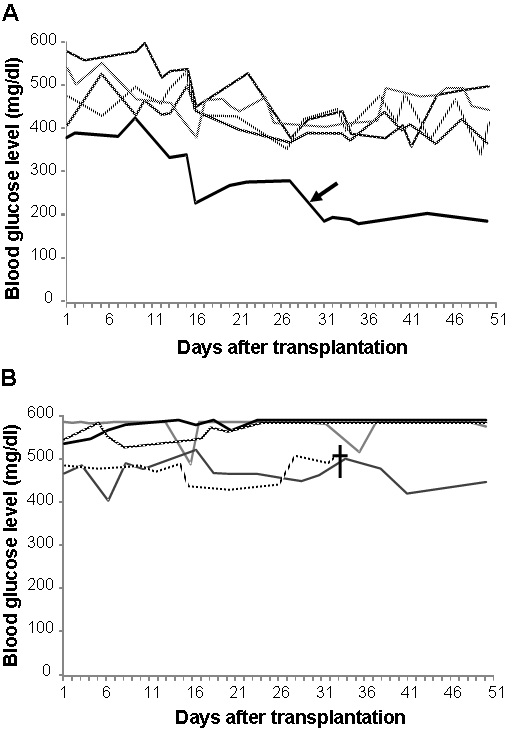 Figure 9: Changes in blood glucose levels measured for diabetic mice into which 700 IEQ’s of spherical tissues generated from the pancreatic tissues of donor mice of the same strain were transplanted under the kidney capsule. Day “1” indicates one day after transplantation. Blood glucose levels above 400mg/dl and below 200mg/dl are considered diabetic and normal, respectively. Transplanted mice showed gradually-decreasing blood glucose levels in the first 4 weeks; one recipient (arrow) reached at normal glucose levels (A). Other transplanted mice showed little or no glucose control (B). One diabetic control mouse (cross) without transplants died during the experiment.
Figure 9: Changes in blood glucose levels measured for diabetic mice into which 700 IEQ’s of spherical tissues generated from the pancreatic tissues of donor mice of the same strain were transplanted under the kidney capsule. Day “1” indicates one day after transplantation. Blood glucose levels above 400mg/dl and below 200mg/dl are considered diabetic and normal, respectively. Transplanted mice showed gradually-decreasing blood glucose levels in the first 4 weeks; one recipient (arrow) reached at normal glucose levels (A). Other transplanted mice showed little or no glucose control (B). One diabetic control mouse (cross) without transplants died during the experiment.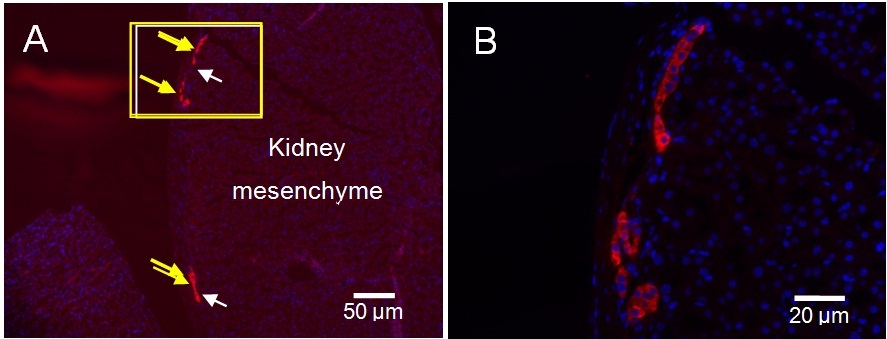
Figure 10: Immunohistological images showing insulin-positive cells beneath a kidney capsule where spherical tissue were transplanted. Arrows indicate the clusters of insulin-positive cells (A), and a rectangular area is magnified (B).
Aldefluor-positive cells’ proliferation in co-culture with stellate cells
Aldefluor-E-cadherin double positive cells sorted constituted a minute population (less than 0.1%) of the total cells in the pancreatic debris (Figure 11), and these cells did not proliferate well in the serum-free medium described earlier. When conditioned media from a separately-cultured stellate cells isolated from the pancreatic debris were added to the serum-free medium, cell proliferation continued for only a few days. In a co-culture environment, where the stellate cells were grown in an insert and loaded into 12 well plates seeded with Aldefluor-E-cadherin double positive cells, the latter proliferated continuously (Figure 12A-E).
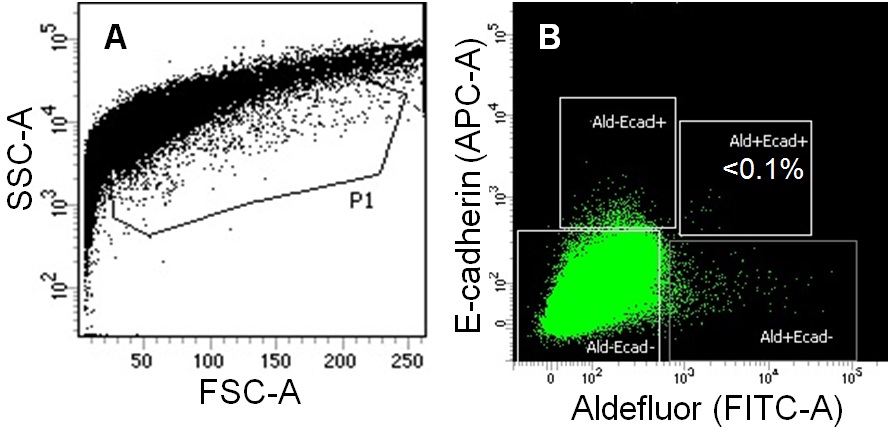 Figure 11: Aldefluor and E-cadherin double positive epithelial cells were sorted from pancreatic debris by FACS. A cell population at a relatively low granulity of the side scatter was gated as P1 (A), from which four subpopulations including the double-positive for Aldefluor and E-cadherin were sorted (B).
Figure 11: Aldefluor and E-cadherin double positive epithelial cells were sorted from pancreatic debris by FACS. A cell population at a relatively low granulity of the side scatter was gated as P1 (A), from which four subpopulations including the double-positive for Aldefluor and E-cadherin were sorted (B).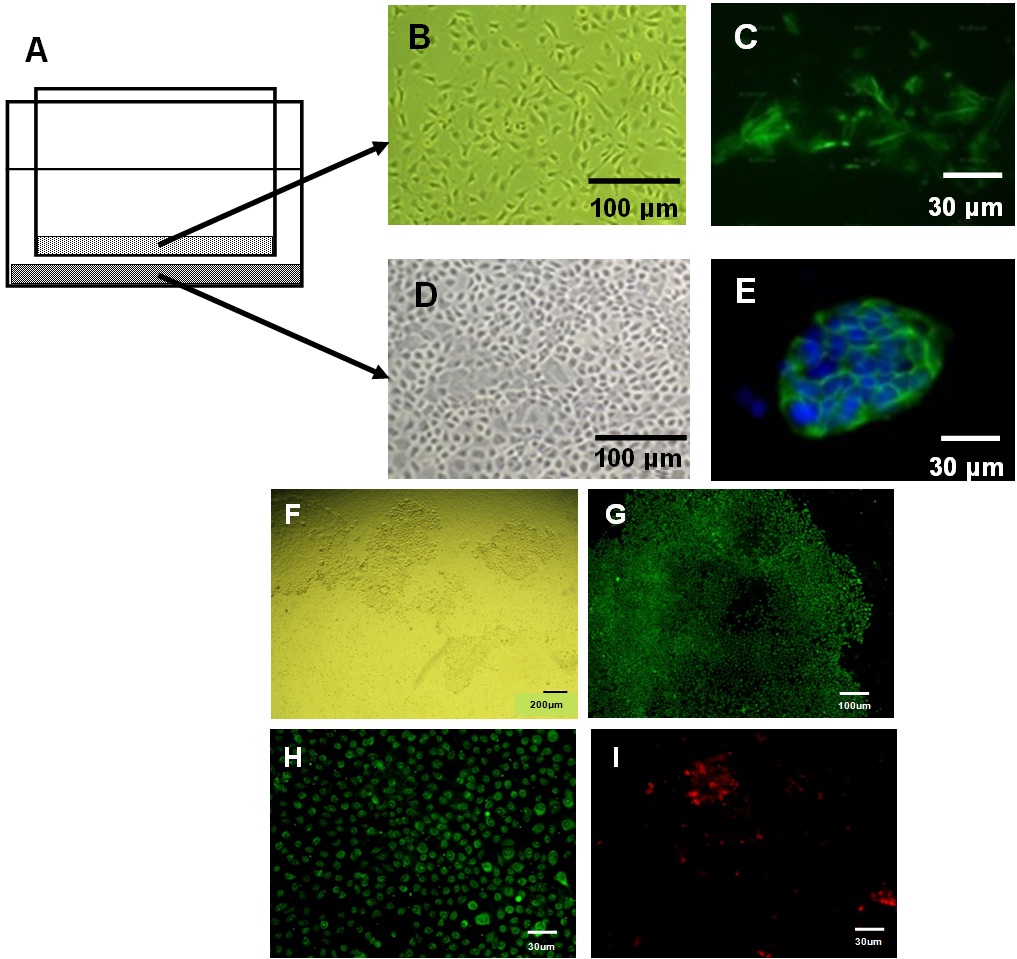
Figure 12: A diagram representing a co-culture of stellate cells housed in an insert is loaded into a transwell culturing an Aldefluor-E-cadherin double positive epithelial cells (A); Phase contrast images of the corresponding stellate (B) and epithelial cells (D), and immunocytochemical images of α-SMA- (C) and E-cadherin-positive cells (E), respectively. Several colonies of the epithelial cells in the transwell (F) co-cultured with stellate cells (A). These epithelial cells cultured in differentiation media contained mostly glucagon-positive cells (G). In a magnified view, among the glucagon-positive cells (H) are small fractions of insulin-positive cells (I).
DISCUSSION
We found that the co-culture of Non-Endocrinal Epithelial Cells (NEECs) and Vascular Endothelial Cells (VECs) both enriched from adult murine pancreas enhanced the generation of islet-like tissues with increased values in differentiation parameters. The co-culture yielded larger amounts of islet-like tissues, greater proportions of insulin-positive cells and the secretion of more C-peptide than the control tissues without VECs. Endothelial cells play important roles in not only supporting the functions of mature pancreatic endocrine cells but also inducing pancreatic progenitors in organogenesis [5,11]. They interact with adjacent endocrine cells by creating a microenvironment called ‘vascular niche’ [20]. At the vascular niche, VECs are known to secrete soluble factors such as HGF to help adjacent endocrine cells for proper maintenance and function [13] as well as extracellular matrices to help endocrine progenitors for proliferation and differentiation [21]. In pancreas, VECs reside in a continuous vascular system that innervates the entire organ from islets to mesenchyme [22]. In late pancreatic organogenesis, when endocrine progenitors bud out from ductal areas and coalesce to form spherical pre-islet structures, endothelial cells form a microvasculature network into the newly-developing islets [23,24]. These various supportive roles of the organ-specific endothelial cells infer their potential to promote even adult pancreatic progenitors in the context of islet regeneration, which has not yet been examined to date. Thus, we tested if VECs isolated from the pancreas could enhance the differentiation of adult pancreatic stem-like cells into endocrinal cells in In Vitro environments.
Both immunocytochemical and mRNA analyses indicated increased expressions of Ngn3 by NEECs when cultured with cyclopamine KAAD and all-trans retinoic acid, which suggests that the heterogeneous populations of NEECs indeed contained stem-like cells with endocrinal differentiation potentials. In synchronized differentiation of ES cells toward pancreatic endocrinal stem cells, Ngn3 expression is known to occur transiently following the down regulation of Notch1 and Hes1 [25,26]. Thus, the concomitant expression of Ngn3 along with that of Notch1 and Hes1 (Figure 4) characterizes the temporal heterogeneity of the NEECs whose subpopulations were undergoing different stages of endocrinal differentiation.
To test if the interaction with VECs would enhance endocrinal differentiation In Vitro, we co-cultured the VECs and NEECs to form a spherical tissue complex in a suspension culture, the method previously used for generating islet-like tissues and known to increase their insulin-secretion ability [27,28]. Cells dissociated from culture containers were swirl-incubated in a culture medium to promote direct homotypic and heterotypic cell-to-cell contacts. Furthermore, in the light of recent finding on ES cell’s differentiation into hepatocytes, swirling motion exerted a certain degree of sheer stress, which in combination with some soluble factors in the medium is known to enhance a differentiation process [29,30].
The spherical tissue complex had increased proportions of insulin-positive cells, amounts of secreted C-peptide, glucose responsiveness, and mRNA expressions of Ins1, Ins2 and glucagon. These data suggest that VECs enhanced the differentiation of progenitors residing in NEECs into functional endocrinal cells. Whether such enhancement was primarily of direct cell-to-cell contact or by means of some extracellular materials secreted by VECs is yet to be clarified. With respect to the contribution of the extracellular materials, it is worth noting that the co-culture increased the amount of tissues generated. Since the amount of VECs mixed with NEECs was less than 30% of the total cell number, the doubled tissue volume as a result of co-culture suggests that VECs had secreted and increased the amount of extracellular materials into the tissue complex.
In vivo evaluation of the tissue complex also showed its overall positive functional qualities in spite of the ambivalent outcomes regarding its In vivo glucose-controlling ability. Among the total of 9 recipient diabetic mice transplanted, only one individual recovered normoglycemia, but notably all of the recipients of the tissue complex survived throughout the experimental period, and a significant number of them had fluctuating blood glucose levels between 300 and 600mg/dl. In contrast, the control diabetic mice without the tissue transplants died during the experimental period.
Aldefluor-E-cadherin double positive pancreatic cells in adult mice have been isolated as a facultative endocrinal stem-like cell population, which required a spheroid culture for continuous clonetic proliferation [4]. We found that these stem-like cells proliferated as a monolayer when co-cultured with stellate cells, suggesting that they require specific cellular niches but are somewhat flexible in choice of physical environments. Since the monolayer NEECs that we have established from pancreatic tissue debris contained some stellate cells, their continuous proliferation possibly entails similar interactions in the co-culture environments.
Our present data indicate that generation of tissue complex of NEECs and VECs both from pancreatic tissue debris can increase its potential of islet-like functions. In Vitro generation of islet-like tissues if performed efficiently will offset the shortage of transplantable donor islets. Once optimized, the tissue complex method will be a feasible application to human tissues for not only therapeutic but also in vitroscreening purposes.
Both immunocytochemical and mRNA analyses indicated increased expressions of Ngn3 by NEECs when cultured with cyclopamine KAAD and all-trans retinoic acid, which suggests that the heterogeneous populations of NEECs indeed contained stem-like cells with endocrinal differentiation potentials. In synchronized differentiation of ES cells toward pancreatic endocrinal stem cells, Ngn3 expression is known to occur transiently following the down regulation of Notch1 and Hes1 [25,26]. Thus, the concomitant expression of Ngn3 along with that of Notch1 and Hes1 (Figure 4) characterizes the temporal heterogeneity of the NEECs whose subpopulations were undergoing different stages of endocrinal differentiation.
To test if the interaction with VECs would enhance endocrinal differentiation In Vitro, we co-cultured the VECs and NEECs to form a spherical tissue complex in a suspension culture, the method previously used for generating islet-like tissues and known to increase their insulin-secretion ability [27,28]. Cells dissociated from culture containers were swirl-incubated in a culture medium to promote direct homotypic and heterotypic cell-to-cell contacts. Furthermore, in the light of recent finding on ES cell’s differentiation into hepatocytes, swirling motion exerted a certain degree of sheer stress, which in combination with some soluble factors in the medium is known to enhance a differentiation process [29,30].
The spherical tissue complex had increased proportions of insulin-positive cells, amounts of secreted C-peptide, glucose responsiveness, and mRNA expressions of Ins1, Ins2 and glucagon. These data suggest that VECs enhanced the differentiation of progenitors residing in NEECs into functional endocrinal cells. Whether such enhancement was primarily of direct cell-to-cell contact or by means of some extracellular materials secreted by VECs is yet to be clarified. With respect to the contribution of the extracellular materials, it is worth noting that the co-culture increased the amount of tissues generated. Since the amount of VECs mixed with NEECs was less than 30% of the total cell number, the doubled tissue volume as a result of co-culture suggests that VECs had secreted and increased the amount of extracellular materials into the tissue complex.
In vivo evaluation of the tissue complex also showed its overall positive functional qualities in spite of the ambivalent outcomes regarding its In vivo glucose-controlling ability. Among the total of 9 recipient diabetic mice transplanted, only one individual recovered normoglycemia, but notably all of the recipients of the tissue complex survived throughout the experimental period, and a significant number of them had fluctuating blood glucose levels between 300 and 600mg/dl. In contrast, the control diabetic mice without the tissue transplants died during the experimental period.
Aldefluor-E-cadherin double positive pancreatic cells in adult mice have been isolated as a facultative endocrinal stem-like cell population, which required a spheroid culture for continuous clonetic proliferation [4]. We found that these stem-like cells proliferated as a monolayer when co-cultured with stellate cells, suggesting that they require specific cellular niches but are somewhat flexible in choice of physical environments. Since the monolayer NEECs that we have established from pancreatic tissue debris contained some stellate cells, their continuous proliferation possibly entails similar interactions in the co-culture environments.
Our present data indicate that generation of tissue complex of NEECs and VECs both from pancreatic tissue debris can increase its potential of islet-like functions. In Vitro generation of islet-like tissues if performed efficiently will offset the shortage of transplantable donor islets. Once optimized, the tissue complex method will be a feasible application to human tissues for not only therapeutic but also in vitroscreening purposes.
ACKNOWLEDGMENTS
Special thanks to Dr. Kanae Mitsunaga at Center for iPS Cell Research and Application for technical assistance in FACS Aria II, and Shinako Kuramoto and Keiko Matsui at Kyoto University Hospital for laboratory assistance. This work was supported by JSPS Grant-in-Aid for Challenging Exploratory Research (Grant No. 24659083).
REFERENCES
- Puri S, Folias AE, Hebrok M (2015) Plasticity and dedifferentiation within the pancreas: development, homeostasis, and disease. Cell Stem Cell 16: 18-31.
- Carpino G, Cardinale V, Gentile R, Onori P, Semeraro R, et al. (2014) Evidence for multipotent endodermal stem/progenitor cell populations in human gallbladder. J Hepatol 60: 1194-1202.
- Wang Y, Lanzoni G, Carpino G, Cui CB, Dominguez-Bendala J, et al. (2013) Biliary tree stem cells, precursors to pancreatic committed progenitors: evidence for possible life-long pancreatic organogenesis. Stem Cells 31: 1966-1979.
- Rovira M, Scott SG, Liss AS, Jensen J, Thayer SP, et al. (2010) Isolation and characterization of centroacinar/terminal ductal progenitor cells in adult mouse pancreas. Proc Natl Acad Sci USA 107: 75-80.
- Lammert E, Cleaver O, Melton D (2001) Induction of pancreatic differentiation by signals from blood vessels. Science 294: 564-567.
- Kim SK, Hebrok M (2001) Intercellular signals regulating pancreas development and function. Genes Dev 15: 111-127.
- Matsumoto K, Yoshitomi H, Rossant J, Zaret KS (2001) Liver organogenesis promoted by endothelial cells prior to vascular function. Science 294: 559-563.
- Cleaver O, Melton DA (2003) Endothelial signaling during development. Nat Med 9: 661-668.
- Crivellato E, Nico B, Ribatti D (2007) Contribution of endothelial cells to organogenesis: a modern reappraisal of an old Aristotelian concept. J Anat 211: 415-427.
- Zaret KS, Grompe M (2008) Generation and regeneration of cells of the liver and pancreas. Science 322: 1490-1494.
- Nikolova G, Jabs N, Konstantinova I, Domogatskaya A, Tryggvason K, et al. (2006) The vascular basement membrane: a niche for insulin gene expression and Beta cell proliferation. Dev Cell 10: 397-405.
- Bahary N, Zon LI (2001) Development. Endothelium--chicken soup for the endoderm. Science 294: 530-531.
- Johansson M, Mattsson G, Andersson A, Jansson L, Carlsson PO (2006) Islet endothelial cells and pancreatic beta-cell proliferation: studies In Vitro and during pregnancy in adult rats. Endocrinology 147: 2315-2324.
- Cano DA, Hebrok M, Zenker M (2007) Pancreatic development and disease. Gastroenterology 132: 745-762.
- Xu X, D'Hoker J, Stangé G, Bonné S, De Leu N, et al. (2008) Beta cells can be generated from endogenous progenitors in injured adult mouse pancreas. Cell 132: 197-207.
- de Koning EJP, Nienaber C, Yatoh S, Patterson D, Rask-Madsen C, et al. (2006) Endothelial cell involvement in proliferation and differentiation of human pancreatic duct cells (Abstract). Diabetologia 49: 290.
- Rivas-Carrillo SD, Kanamune J, Iwanaga Y, Uemoto S, Daneri-Navarro A, et al. (2011) Endothelial cells promote pancreatic stem cell activation during islet regeneration in mice. Transplant Proc 43: 3209-3211.
- Erkan M, Adler G, Apte MV, Bachem MG, Buchholz M, et al. (2012) StellaTUM: current consensus and discussion on pancreatic stellate cell research. Gut 61: 172-178.
- Bachem MG, Schunemann M, Ramadani M, Siech M, Beger H, et al. (2005) Pancreatic carcinoma cells induce fibrosis by stimulating proliferation and matrix synthesis of stellate cells. Gastroenterology 128: 907-921.
- Nikolova G, Strilic B, Lammert E (2007) The vascular niche and its basement membrane. Trends Cell Biol 17: 19-25.
- Cirulli V, Beattie GM, Klier G, Ellisman M, Ricordi C, et al. (2000) Expression and function of alpha(v)beta(3) and alpha(v)beta(5) integrins in the developing pancreas: roles in the adhesion and migration of putative endocrine progenitor cells. J Cell Biol 150: 1445-1460.
- Menger MD, Vajkoczy P, Beger C, Messmer K (1994) Orientation of microvascular blood flow in pancreatic islet isografts. J Clin Invest 93: 2280-2285.
- Piper K, Brickwood S, Turnpenny LW, Cameron IT, Ball SG, et al. (2004) Beta cell differentiation during early human pancreas development. J Endocrinol 181: 11-23.
- Liew CG, Andrews PW (2008) Stem cell therapy to treat diabetes mellitus. Rev Diabet Stud 5: 203-219.
- Jensen J, Heller RS, Funder-Nielsen T, Pedersen EE, Lindsell C, et al. (2000) Independent development of pancreatic alpha- and beta-cells from neurogenin3-expressing precursors: a role for the notch pathway in repression of premature differentiation. Diabetes 49: 163-176.
- Jensen J, Pedersen EE, Galante P, Hald J, Heller RS, et al. (2000) Control of endodermal endocrine development by Hes-1. Nat Genet 24: 36-44.
- Ogata T, Park KY, Seno M, Kojima I (2004) Reversal of streptozotocin-induced hyperglycemia by transplantation of pseudoislets consisting of beta cells derived from ductal cells. Endocr J 51: 381-386.
- Todorov I, Omori K, Pascual M, Rawson J, Nair I, et al. (2006) Generation of human islets through expansion and differentiation of non-islet pancreatic cells discarded (pancreatic discard) after islet isolation. Pancreas 32: 130-138.
- Zhang S, Zhang Y, Chen L, Liu T, Li Y, et al. (2013) Efficient large-scale generation of functional hepatocytes from mouse embryonic stem cells grown in a rotating bioreactor with exogenous growth factors and hormones. Stem Cell Res Ther 4: 145.
- Sasaki K (2014) Large-scale generation of differentiated cells to achieve regenerative medicine. Stem Cell Res Ther 5: 10.
Citation: Kanamune J, Iwanaga Y, Rivas-Carrillo JD, Sumi S, Uemoto S (2015) Tissue Complex of Adult Pancreatic Duct and Vascular Endothelial Cells Promotes In Vitro Differentiation into Insulin-Producing Cells. J Stem Cell Res Dev Ther 2: 005.
Copyright: © 2015 Jun Kanamune, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Journal Highlights
© 2026, Copyrights Herald Scholarly Open Access. All Rights Reserved!