Why Every Year Several New Cancer Drugs Go to Market but There is no Cure Medicine for Alzheimer's? are We Off-Target?
*Corresponding Author(s):
Majid VahedPharmaceutical Sciences Research Center, Shahid Beheshti University Of Medical Sciences, Niayesh Highway, Valiasr Ave, Tehran, Iran, Islamic Republic Of
Email:pc.vahed@gmail.com
SHORT COMMENTARY
Alzheimer's disease (AD) commonly used mouse models. Several monoclonal antibodies have since been shown to clear plaques in mouse models but all have failed to provide benefit to people with AD in trials. The amyloid-β (Aβ) peptides aggregate into plaques contribute to the pathogenesis of AD. There are currently more than 400 trials in people of potential treatments for around the world, but almost no drugs have been brought to the market. Common mouse models of Alzheimer disease. Aggregational and conformational changes of human Aβ peptides into unique S-shaped β-sheet misfolded proteins is a pathological hallmark of neurodegenerative diseases such as AD [1]. The formation of a GM1 cluster was suggested to be cholesterol-dependent [2,3]. It was demonstrated by Matsuzaki that a GM1-including membrane not only enhanced the aggregation of Aβ but also converted amyloid peptides into toxic fibrils that showed high hydrophobicity and contained anti-parallel β-sheets [4] (Figure 1). Residue Arg5 (Figure 2) was observed to play a key role for Aβ stable binding to the membrane in the simulations [1]. Three amino acid differences distinguish Aβ from its human homolog (hAβ) and mice (mAβ) which are located within the N-terminal domain (Figure 1; Arg-5→Gly, Tyr-10→Phe, and His-13→Arg) ; however Arg-5→Gly amino acid differences hallmark for Aβ stable binding to the membrane. Furthermore, the residue 13 of mAβ and rAβ are Arg rather than His in hAβ; these mutations are critical in AD [5].

Figure 1: Amino acid sequences of hAβ (1-42), mAβ (1-42), and rAβ (1-42). Amino acids with positive and negative charges are shown in blue and red, respectively. His residues represented in green have no charge at physiological pH.
The principal component of extracellular plaque aggregation in the brain has been reported to be stabilized Fe2+ and Fe3+ ions that promote the formation of Aβs due to free radicals that could cause the death of neurons by apoptosis [6,7]. Interestingly mole-rat brains of around 32 years of age, had levels of rAβ similar to AD models but not observed any AD symptom [8]. Experimental studies suggest that Aβ fibrillation (main-pathway is known to be more toxic) and amorphous (off-pathway) formations are two competitive pathways influencing by factors such as metal binding, pH and temperature [9]. The hAβ peptides have a different net charge, which contains 9 polar residues; the hAβ peptides have 11 polar residues and rAβ, which contains 10 polar residues [10]. Positive charges substitution of histidine (His13) to Arg13 is represented in mAβ and rAβ. Furthermore, in mAβ with glycine five (Gly5) to Arg5, the replacement has one more positively charged residue. At pH 7, the net charges were -3 for hAβ, -2 for rAβ, and -1 for mAβ. In our previous study, hAβ and rAβ have different behaviors in the initial stage of structural transformation of Aβ42 peptides in the presence of Fe2+ and Fe3+. We reported that the intermolecular an important feature that intramolecular C-terminal β-sheets can play key roles in Aβs main and off pathway [2]. Fe2+ and Fe3+ play key roles in folding Aβs [5]. Fe3+ ions stabilized the folding of β-sheets on the C-terminal with the “Intramolecular tailoring approach” by binding iron to the Glu22 and the carboxyl group of Ala42 [5]. Saccharomyces cerevisiae has high similarity in nucleic acid, and amino acid sequencing with mammalian might be a good model for substitutions with mice.
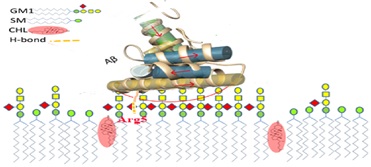
Figure 2: View of the equilibrated mixed membrane of a lipid composition of GM1 ganglioside, sphingomyelin (SM), and cholesterol (CHL). Interaction of three molecules in the hAβ complex, extracted from the Last snapshot structure of the MD simulation for the model with Five Aβs. Critically stable anti-parallel β-sheet structure was formed between the C-terminals of two Aβs.
CONCLUSION
The most common forms of toxic hAβ contained anti-parallel β-sheets observed in human which formed on the GM1-including membrane. Arg5 was observed to play a key role for Aβ stable binding to the membrane. Therefore, When compared to hAβ, mAβ peptides were found residue 5 of mAβ42 is Gly instead of Arg of hAβ which in the presence of a GM1-containing membrane, hAβ was aggregated to be a toxic form, but mAβ developed into a different form and the aggregates was less toxic. This is not surprising, as there are no suitable ligands for the Fe of heme in biology. Imidazoles are representative of the heme group and side chain of histidine amino acids. Therefore, hAβ with three his residues can mimic heme groups in interaction with Fe ions. Therefore, by binding iron, the conformational flexibility of hAβ peptides with three His is lesser than mAβ and rAβ with two His. To sum it up, the feature of hAβ is critical in AD, and mAβ and rAβ with different properties might not be a good model for AD.
REFERENCES
- Vahed M, Neya S, Matsuzaki K, Hoshino T (2018) Simulation study on complex conformations of Aβ42 peptides on a GM1 ganglioside containing lipid membrane. Chem Pharm Bull 66: 170-177.
- Vahed M, Neya S, Matsuzaki K, Hoshino T (2018) Analysis of Physicochemical Interaction of Aβ40with GM1 Ganglioside-Containing Lipid Membrane. J Phys Chem B 122: 3771-3781.
- Kakio A, Nishimoto SI, Yanagisawa K, Kozutsumi Y, Matsuzaki K (2001) Cholesterol-dependent formation of GM1 ganglioside-bound amyloid beta-protein, an endogenous seed for Alzheimer amyloid. J Biol Chem 276: 24985-24990.
- Matsuzaki K (2014) How do membranes initiate Alzheimer's Disease? Formation of toxic amyloid fibrils by the amyloid β-protein on ganglioside clusters. Acc Chem Res 47: 2397-2404.
- Vahed M, Sweeney A, Shirasawa H, Vahed M (2019) The initial stage of structural transformation of Aβ42 peptides from the human and mole rat in the presence of Fe2+ and Fe3+: Related to Alzheimer's Comput Biol Chem 83: 107128.
- Greenough MA, Camakaris J, Bush AI (2013) Metal dyshomeostasis and oxidative stress in Alzheimer’s disease. Neurochem Int 62: 540-555.
- Everett J, Céspedes E, Shelford LR, Exley C, Collingwood JF, et al. (2014) Evidence of redox-activeiron formation following aggregation of ferrihydriteand the Alzheimer’s disease peptideb-amyloid. Inorg Chem 17: 2803-2809.
- Edrey YH, Medina DX, Gaczynska M, Osmulski PA, Oddo S, et al. (2013) Amyloid beta and the longest-lived rodent: The naked mole-rat as a model for natural protection from Alzheimer’s Neurobiol Aging 34: 2352-2360.
- Jiang D, Rauda I, Han S, Chen S, Zhou F (2012) Aggregation pathways of the amyloid β (142) peptide depend on its colloidal stability and ordered beta-sheet stacking. Langmuir 28: 12711-12721.
- Vahed M, Ahmadian G, Ameri N, Vahed M (2019) G-rich VEGF aptamer as a potential inhibitor of chitin trafficking signal in emerging opportunistic yeast infection. Comput Biol Chem 80: 168-176.
Citation: Vahed M, Esmaeili R, Vahed M (2019) Why Every Year Several New Cancer Drugs Go to Market but There is no Cure Medicine for Alzheimer's? are We Off-Target? J Alzheimers Neurodegener Dis 5: 032.
Copyright: © 2019 Mohammad Vahed, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.