The Acute Porphyric Attack: A Difficult Diagnosis for a Potential Lethal Event in Emergency Medicine
*Corresponding Author(s):
Paolo VenturaDepartment Of Medical And Surgical Science For Children And Adults, Centre For Porphyrias - Division Of Internal Medicine II, Policlinico Hospital Of Modena, University Of Modena And Reggio Emilia, Largo Del Pozzo 71, 41124 Modena MO, Italy
Tel:+39 594222807,
Fax:+39 594224363
Email:paoloven@unimore.it
Abstract
The porphyrias are a heterogeneous group of metabolic disorders due to an inherited (but in some forms the disturbance may also be acquired) enzymatic deficiency in the metabolic pathway of heme biosynthesis. The variable degree of block in the heme biosynthetic pathway due to the enzyme deficiency results in accumulation of different metabolic intermediates, whose toxicity is responsible for the peculiar (cutaneous and/or neurovisceral) clinical pictures observed in each of these diseases. According to the clinical features, the porphyrias are classified as “acute” (or neuropsychic) [characterized by acute neurovisceral crises (the acute porphyric attack) involving the autonomic and/or central nervous system, but also the liver and the kidney] and “on acute” (or dermatological) (mostly presenting with cutaneous lesions, due to photosensitivity).
The acute porphyrias are often misdiagnosed diseases: the acute porphyric attack may in fact mimic many other more common medical and neuropsychiatric conditions; its delayed diagnosis and treatment (or its inappropriate treatment) may result in a fatal outcome. For these reasons, many different specialists, such as surgeons, psychiatrists, gastroenterologists, neurologists, emergency physicians and dermatologists may be variably involved in the diagnostic process, especially in those cases presenting with acute and life-threatening clinical features. An early and definitive diagnosis is mandatory to improve outcomes and to assure that potentially harmful drugs are avoided. To date, the availability of an adequate treatment has significantly improved the outcome of the acute porphyric attacks, so the knowledge about the management of these events may be relevant for the physicians working in internal and emergency medicine units.
Keywords
INTRODUCTION
The porphyrias are a heterogeneous group of rare metabolic diseases resulting from an inherited (in some circumstances the disturbance may be acquired) enzymatic deficiency in the pathway of heme biosynthesis (Figure 1). The enzymatic defect gives rise to a sort of metabolic ‘bottleneck’ in the biosynthetic pathway, which may result in an accumulation of metabolic intermediates (porphyrins and/or non porphyrin precursors) whose toxicity is responsible for the peculiar clinical pictures of these diseases [1-4]. In general, the porphyrias are classified as erythropoietic or hepatic, depending on the tissue where the metabolic defect is prevalent and thus on the site of major production of porphyrins and/or their precursors. These diseases are also classified as acute or non-acute, according to their main clinical features, even though the clinical manifestations may overlap in some forms [2,4-6].
 Figure 1:Biosynthesis of Heme (enzymes and genes involved and correlated diseases are reported).
Figure 1:Biosynthesis of Heme (enzymes and genes involved and correlated diseases are reported).
The clinical landmarks of the acute porphyrias are recurrent acute crisis (acute porphyric attack-APAs), characterized by a severe neuro-visceral involvement, expressing with abdominal pain, mental symptoms and potentially life-threatening acute neuropathy [2,5,7-11]. The non-acute porphyrias are characterized mainly by cutaneous manifestations related to photosensitivity (Table 1) [1,2,4,12-21]. It is to be remarked as all “acute” porphyrias are “hepatic” porphyrias, this suggesting the key-role of the liver in the pathogenesis of these diseases [6,7,15].
| Acute Porphyrias |
|
ALA dehydratase- deficiency Porphyria(ALAD-P) |
| Non-Acute (Cutaneous) Porphyrias |
|
Porphyria Cutanea Tarda (PCT) |
The clinical features alone are not so specific and suitable to confirm a diagnosis of APA. For this reason, the knowledge and the correct interpretation of the appropriate tests are mandatory for an accurate clinical management of these diseases: a delayed diagnosis and/or an inappropriate treatment of an APA may be in fact fatal [1,6,8,22-25]. In last decades, the availability of effective therapies (infusion-stable heme preparations) has significantly improved the outcome of APAs: for this reason, the knowledge about the diagnostic steps and the clinical management of these diseases is relevant, especially for the physicians working in internal and emergency medicine units [22,25-27].
In this review, we propose some suggestions for diagnosing and managing an acute porphyric attack based on literature reviews and clinical experience of our centre, with long-time expertise in the clinical management of patients affected by porphyrias.
MATERIAL AND METHODS
Studies and review articles related to Acute Porphyric Attack (APA) we referred in this review were sought via electronic databases (PubMed®, Medline®, Embase®) and were identified from key references within articles. Search terms and MeSH headings we used included the word crisis, acute attack, drugs, precipitating factors combined with each of the following: porphyrias, ala-synthase, haem, heme, haematin, haem arginate, acute intermittent porphyria, variegate porphyria, hereditary coproporphyria, ala-d deficiency porphyria, lead poisoning. Due to the matter of content, no formal evaluation of level of evidence was conducted in developing this narrative review.
THE ACUTE PORPHYRIAS: GENERAL ASPECTS AND NOTICES OF EPIDEMIOLOGY
Heme synthesis proceeds through different and complex biochemical reactions, in turn catalysed by specific enzymes: succinyl-coenzyme A and glycine are progressively converted into the complex tetrapyrrole ring of protoporphyrin IX that, after complexation with an atom of Fe2++, leads to the formation of heme (Figure 1). A disturbance in the activity of each of these enzymes causes accumulation of different patterns of precursors, whose toxic effects are responsible for the different clinical pictures characterizing the porphyrias [1,3]. Four kinds of “porphyrias” may present with recurrent attacks of neuro-visceral symptoms (Acute Porphyric Attack- APA): for this reason they are also classified as “acute porphyrias” [6,7]. The most common of acute porphyrias is Acute Intermittent Porphyria (AIP) [28-30]. It has been reported that APAs characterizing AIP are clinically more severe, even though they are indistinguishable from those characterizing the other less common variants of acute porphyrias: Variegate Porphyria (VP) [3,31-34], Hereditary Coproporphyria (HCP) [3,5,35,36], and porphyria due to ALA dehydratase deficiency (ALAd-P) (which is extremely rare) [37]. Very similar clinical severe manifestations may also occur in case of lead poisoning, a condition also referred as plumboporphyria: it may be considered as an acquired disturbance of heme metabolism, being caused by the inhibition of ALA-dehydratase by lead [38,39].
A reliable epidemiologic data collection about acute porphyrias is difficult due their low clinical penetrance: it is thought that less than 10-15% (only about 1-3%, according to most recent data) of carriers of the enzymatic defect develops overt APAs [40-42]. Statistics about morbidity of acute porphyrias derive mostly from the experience of specialist porphyria centers [1,30], and from some systematic studies concerning each kind of porphyria from different countries [33,43-50]. The most recent prospective survey reports a similar incidence for AIP in all European countries [42] (ranging from 0.11 to 0.22 per year per million, with an overall incidence of 0.12), with the significant exception of Sweden, where the incidence of about four-fold higher (0.51 per year per million) is probably explained by a “founder effect” [46]. For VP (about half that of AIP) and HCP (0.2 per year per 10 millions), the incidence in Europe is lower. In this study, the calculated overall prevalence of AIP in Europe (including Sweden) is about 5.9 per million inhabitants; it resulted significantly lower with respect to previous estimates (from 10-20 per million to 101 per million), where probably all subjects with AIP, even those who never experienced symptoms, have been included [1,30,49,51]. Similar considerations are valid for the calculated European prevalence of VP (3.2 cases for million inhabitants) [42]; in South Africa, where a “founder effect” has been well-demonstrated, the genetic defect responsible for VP has an exceptionally high frequency (1:300) [52,53].
To this regard, some retrospective studies have showed that in the last 50 years the number of symptomatic AIP patients (i.e. patients experiencing APAs) declined with time and this trend seemed to have continued subsequently [47,54]; similarly, a decrease in APA frequency in VP has been noted in South Africa [55]. Thus, this observed lower prevalence may also be consistent with a decreasing incidence of new acute attacks over the past decades, which could be explained by a significant improvement in diagnosis, treatment, family screening and preventive counseling.
In old studies, mortality during an APA has been reported to be as high as 50- 60% [56]. With modern treatment, however, APAs have rarely a lethal outcome. Nevertheless, according to an American report, the mortality rate was three times higher among patients with AIP, as compared to the general population, being symptoms associated with the APA itself the major cause of this increase in mortality [57].
THE ACUTE PORPHYRIC ATTACK: CLINICAL PICTURES
The clinical landmark of an acute porphyria is the Acute Porphyric Attack (APA). APA is characterized mostly by (recurrent) acute crisis with a severe neuro-visceral involvement, whose clinical expression may be greatly variable and able to mimic many other diseases (Table 2); for this reason, the acute porphyrias are often misdiagnosed diseases and many different specialists, such as surgeons, psychiatrists, gastroenterologists, neurologists and emergency physicians may be involved in the diagnostic process, especially in the cases presenting with very severe and life-threatening symptoms [58].
| Signs/symptoms of APA | % | Conditions mimicked by APA | |||||
| Surgical Conditions |
Hematological conditions |
Gastroenterological conditions | Cardiovascular conditions | Dismetabolic/ Disendocrine conditions |
Neurologic and psychyatric conditions |
||
| (Severe) abdominal pain | 95-97 | -Peritonitis -Appendicitis -Acute pancreatitis and cholecystitis -Intestinal ischemia |
-Acute hemolytic crisis -Acute drepanocytic crisis |
-Paralytic Ileus -Acute pancreatitis -Acute cholecystitis |
|||
| Nausea, Vomiting Constipation |
48-85 46-52 |
-Acute gastroenteritis with vomiting | |||||
| Tachycardia Hypertension (diastolic>85 mmHg) Chest pain |
65-80 38-64 8-15 |
-Hypertensive crisis -Tachyarrhythmia -Acute Coronary Syndrome |
-Pheocromocytoma | ||||
| Hypotension Hyponatriemia (<120 mEq/L) |
15-22 25-35 |
-Acute hypoadrenalism (Addisonian crisis) -SIADH |
|||||
| Peripheral motor -neuropathy Sensory neuropathy Hypo/areflexia Back pain Seizures Coma |
40-60 20-28 20-30 20-30 10-20 2-10 |
-Acute hypoparathyroidism (hypocalcemic crisis) -Acute hyperparathyroidism and other hypercalcemic conditions |
-Guillain–Barre` syndrome -Idiopathic/ metabolic autoimmune -Polineuropathies -Emicrania -Epilepsy -Acute myopathies |
||||
| Mental changes/psychosis | 10-40 | -Acute psychotic attack -Delirium -Acute panic attack |
|||||
An APA may be preceded by a period of minor behavioral changes such as restlessness, insomnia, anxiety and irritability: these symptoms may evolve rapidly to a severe autonomic and acute motor and sensory neuropathy. APAs may develop over hours or days and can last up to several weeks. The most common symptom of an APA is the onset of severe abdominal pain, usually described as excruciating by the patients and often suggesting an “acute abdomen” nevertheless the symptoms typically appear disproportionate with respect to the physical examination (abdominal tenderness and main peritoneal signs are in fact usually absent). Back pain, often extending to the proximal limbs is also frequently present. The abdominal complaints are generally accompanied by gastroenteric (mostly nausea and vomiting, but also bowel distention, that may develop as an effect of paralytic ileus) and neurological and/or psychiatric symptoms. All components of the peripheral and of the central nervous system may be involved. A motor neuropathy, particularly a proximal motor neuropathy, is quite common in case of severe and prolonged attacks. Muscular weakness usually begins in the extremities of the limbs but can involve any motor neuron or cranial nerve: it can proceed to tetraplegia (resembling a Guillain–Barré syndrome). A bulbar involvement can proceed to respiratory failure [23,58]. Different grade of vegetative dysfunction (systemic arterial hypertension with postural arterial hypotension, tachycardia and constipation) may be also present [1,2].
The involvement of central nervous system may be responsible for seizures and/or mental disturbances (varying from apathy and depression to extreme agitation, mimicking a psychotic attack with hallucinations) [10,11,59-63].
Hyponatremia and hypomagnesemia may occur as a result of dehydration, nephrotoxicity, or also as an effect of excessive release of Antidiuretic Hormone (ADH) and/or inadequate endovenous administration of 10% or 20% glucose (or dextrose) solutions, a standard first-line therapy for APA [64]. These water/electrolyte disturbances may contribute to the neurological and psychiatric symptoms of the APA. During an APA, urine appears often red or reddish-brown, as a consequence of massive excretion of porphyrins [6,58,65,66].
In VP and HCP, a history (or the presence) of a photocutaneous syndrome, characterized by fragile skin and bullous eruptions exclusively interesting the sun-exposed areas (the lesions typically occur on the dorsal aspects of the hands and forearms, the face, ears, and neck), may be present [3,58].
APAs have been rarely described before puberty, with a peak age of presentation in the early 30s, and they are four-to-five times more common in women than in men [2,3,67]. In most people, the disease may remain latent throughout life even in the presence of precipitating factors: it has been estimated that only 10-15% (according to more recent data less than 3%) of gene carriers of an acute porphyria may experience an APA throughout their lifetime [6,41,50,66,68,69]. At least one third of these symptomatic patients have often no family history, this suggesting a condition remained latent or unidentified for several generations. Also the frequency and severity of APAs have a great variability: some patients experience frequent, severe and sometimes life-threatening attacks, even in the absence of exogenous precipitating factors [6]. Nevertheless, in most cases, an APA is precipitated by one or more “triggering factors”, such as drugs, changes in hormone balance (as during menses or hormonal therapies), local or general anesthesia, sedative use (especially barbiturates), misuse of alcohol or illicit substances (amphetamines, cocaine and other derivatives), prolonged fasting or diet restrictions, intense mental or physical stress, or acute infections [1,2,6,70]. In women, recurrent attacks often coincide with the luteal phase of the menstrual cycle [71]. APAs have been described also during pregnancy, coinciding with high estrogen concentrations: for this reason, the symptomatic patients should be advised to avoid pregnancy until remission has been present for at least 2 years; nevertheless in most cases, pregnancy is symptom-free. On the contrary, particular attention should be payed at the delivery and post-delivery periods: they represent in fact important precipitant conditions (stress, blood losses, starvation, etc.) and a high frequency of APAs has been described throughout. If APAs do occur during pregnancy or in the immediate post-delivery period, they should be treated as is customary [72,73].
THE ACUTE PORPHYRIC ATTACK: ASSOCIATED PATHOLOGIC CONDITIONS
Different epidemiological studies have suggested a significant correlation between AIP, arterial systemic hypertension and kidney disease. Hypertension has often been described as well in HCP and VP, but the incidence of renal involvement in these conditions remains ill-defined with respect to that observed in AIP. The treatment of arterial hypertension decreases the risk for renal impairment in carriers of a gene mutation consistent with a form of acute porphyria; symptomatic patients should carefully be managed through regular monitoring of blood pressure and renal function [74,75]. Severe and recurrent motor involvement during APAs may lead to persistent muscle weakness and atrophy [10]. A significant association between acute porphyrias and Hepatocellular Carcinoma (HCC) has also been described: 27% of patients carrying the W198X mutation of the porphobilinogen-deaminase gene are reported to be affected by HCC in a mortality study [76,77]. Cirrhosis, hepatocellular carcinoma, systemic arterial hypertension, and renal impairment have been reported as more common after middle age in AIP and possibly also in VP and HCP, especially in symptomatic patients [78].
THE ACUTE PORPHYRIC ATTACK: PHYSIOPATHOLOGY AND PRECIPITATING FACTORS
The pathophysiologic mechanisms underlying the clinical features of APAs are still poorly understood; possible mechanisms include:
(a) Damage by free radicals formed as a consequence of porphyrin and/or porphyrin praecursors accumulation (not completely confirmed) [79].
(b) A direct neurotoxicity of the accumulating porphyrin precursor Delta-Aminolevulinic Acid (ALA) [80-82].
(c) An acute heme deficiency in nervous and other tissues [83,84].
It has been demonstrated that ALA is neurotoxic [82,85]. The blood-brain barrier protects the brain from toxic agents, but certain areas such as the hypothalamus and limbic area do not have such protection [10]. Moreover, circulating porphyrins and their precursors can cause vascular injury, leading to impaired permeability, and resulting in reversible focal edema in the brain [10]. The autonomic and peripheral nervous systems are especially vulnerable to this toxic action, due to absence of a specific barrier protection. Vulnerability of neurons to toxic reactions seems to be highly variable among individuals: this may explain why some patients are more prone to paresis than others are. Some patients affected by acute porphyria have constantly elevated levels of porphyrins and their precursors also during the symptom-free phase, but even their excretion level increases during an acute attack [65,68]. ALA has also been suggested as a carcinogenic for the liver [76]; it can cross the placenta and possibly cause toxicity to the developing fetal brain [73].
From a biochemical point of view, the landmark of the APA is the significant accumulation of the non-porphyrin precursors Delta-Aminolevulinic Acid (ALA) and Porphobilinogen (PBG) in biological fluids, as an effect of their excessive liver production due to induction of heme biosynthesis [2,3]. The synthetic rate of heme depends mostly on the current activity of the first enzyme of the chain, 5-Aminolevulinate Synthetase (ALA-S1) (a rate-limiting step) (Figure 1). ALA-S1 activity is feedback-governed by the amount of free heme present within the cell (the hepatocyte). A decrease in the availability of this “regulatory” free heme pool gives rise to ALA-S1 induction, and hence to an increase of heme biosynthesis [86]. A second rate-limiting enzymatic reaction is the third step of heme synthesis [catalyzed by Porphobilinogen Deaminase (PBG-D)]. In contrast to ALA-S step, which represents a variable control step for the synthetic process, the constantly limited capacity of the PBGD (HBMS) step functions as a sort of “inbuilt safety-valve” regulating the overproduction of toxic porphyrins along the biosynthetic chain. In the acute porphyrias, the enzymatic activity of PBGD (HBMS) is significantly compromised:
1) As a consequence of a genetically determined deficiency in case of AIP;
2) Probably as an inhibitory effect by porphyrins produced in surplus in case of VP and HCP.
The restoration of a normal porphyrin metabolism in an AIP symptomatic patient (who thereafter became attack-free) after liver transplantation, confirms the key role of the liver as a source of non-porphyrin precursors in acute porphyrias [87,88]. Exogenous and endogenous factors able to induce the expression and/or the activity of ALA-S1 (as in the case of drugs or pathological conditions reducing the free heme pool) are well-known potential triggering factors of APAs [1-3,70].
THE ACUTE PORPHYRIC ATTACK: DIAGNOSIS AND DIFFERENTIAL DIAGNOSIS
The correct and prompt diagnosis of an APA greatly depends on the awareness, the knowledge and experience of the clinician: misdiagnosis is common, because the APAs may be easily confused with other causes of acute abdomen (sometimes leading to unnecessary surgery) or with primary neurologic or mental diseases. The probability of APA being present may be higher when the patient can report that he/she is a member of porphyria kindred, or he/she is actually a carrier of genetic mutations consistent with acute porphyria, in the best case verified by a personal warning card, issued by the specialist responsible for the diagnosis [89]. Emission of dark (red or reddish brown) urine, in correspondence of symptoms onset, is a cardinal sign and it is often present during APA [58,90-92].
As already remarked above, it should be noted that universal signs or symptoms of APA do not exist, and in up to 5%-10% of patients the disease may express without the most widespread and shared features (such as the severe abdominal pain) [59,93]. A family history may be relevant in the presence of symptomatic relatives, but it may also be inconclusive as the acute porphyrias are diseases characterized by highly incomplete penetrance and most carriers may remain life-long asymptomatic. On the other hand, it is recommendable to test immediately patients with abdominal pain and/or other suggestive findings of APA and a family history of acute porphyria [6,58,90,92]. When a patient known to be affected by acute porphyria shows symptoms consistent with APA, the further question is whether the clinical picture is due to its disease or not: not all symptoms in porphyric patients are due to APA; moreover, patients with acute porphyrias may be suffering from other (and more common) diseases. Table 2 summarizes some of the most common pathological conditions whose differential diagnosis should include an APA.
Due to their non-specificity, the clinical features alone are not sufficient either to confirm a diagnosis of APA or to differentiate between the different forms of acute porphyria. For this reason, an immediate (we recommend simultaneously at the onset of symptoms) assessment and interpretation of some appropriate laboratory biochemical tests (i.e. determination and quantification of porphyrins and non-porphyrin precursor patterns in biological samples) are mandatory for an accurate diagnosis, and for starting an appropriate clinical management [3,25,58,90-92,94,95]. According to current knowledge and consensus, an APA is invariably associated with an increased urinary excretion of non-porphyrin precursors [ALA and PBG] [6,25,92,96,97] (Figure 2). For this reason, in case of suspected APA, a fresh light-protected urine sample should be assessed for ALA and PBG concentrations [to date, HPLC assays are the most accurate, but rapid, ion-exchange column tests (column-chromatographic screening tests) are also available] (first-line test) [66,98-102]. In case of significant renal dysfunction, ALA and PBG levels should be assessed in serum [102].
Different Ehrlich’s reagent-based tests (such as the Watson-Schwartz test or the Hoesch test, where the colourless pyrrole PBG forms a red-violet pigment after reaction with p-dimethylaminobenzaldehyde) may be used as a rapid assay (“bed-side” assessment) to test the presence of urinary PBG (qualitative test) [103,104]. These tests may be considered a “first-line” guide to confirm (or to rule-out) a suspicion of APA, if made at or near the time of the clinical onset in case of the most common acute porphyrias (AIP, PV and HCP). They may miss the diagnosis in some extremely uncommon circumstances (false positive and/or negative results):
a) Subjects affected by the rare ALAd-D porphyria or by lead-poisoning, two disorders being characterized by accumulation of ALA, but not of PBG;
b) Subjects immediately treated with heme arginate (which rapidly decreases ALA and PBG);
c) In some cases of HCP and VP, where the increase in ALA and PBG levels may be more transient, as well the corresponding symptoms;
d) In cases of high urinary bilinogen excretion (due to possible cross-reaction with p-dimethylaminobenzaldehyde) [97,105].
During an APA, especially in case of AIP, urinary ALA and PBG are generally very high, so that potential differences in reference ranges among different laboratories are of little relevance, as well the collection of urine samples for 24 hours, which may contribute to delay the diagnosis. The classical 24-hour urine collection approach has been recently replaced by the assessment on spot urine sample, in which the ALA and PBG values are normalized on urinary creatinine. In the presence of suspected symptoms, PBG and ALA levels higher than 5 times the normal range indicate an APA [58,90,106]. It should be recommended that all major medical facilities and emergency units are provided at least for in-house rapid determination of urinary PBG levels [preferably by using rapid (“bedside”) test kits], because a significant delay in diagnosis may be responsible for potential fatal consequences of delayed treatments. If urinary PBG levels are increased, further testing will determine the definite disorder of heme metabolism, although treatment (which is the same regardless of the type of acute porphyria) should never be delayed, waiting for these results [58]. If only the ALA level is substantially increased, ALA-dehydratase porphyria and other causes of ALA-dehydratase deficiency, such as lead poisoning (plumboporphyria) or hereditary tyrosinemia type 1 should be taken into account before starting treatment [3,7,92,97,107,108]. If urinary PBG and ALA levels are normal, an alternative diagnosis to APA must be considered. In our experience, urinary ALA and PBG values significantly decrease with clinical improvement and dramatically after therapy (especially after heme arginate infusion) (Figure 2). Urinary ALA and PBG levels are generally less markedly increased in APAs due to HCP and VP and they decrease more rapidly after an acute attack than in AIP [34,58]. Because treatment of APA does not depend on the type of acute porphyria, the identification of the specific kind of acute porphyria is important mainly for a correct diagnosis, for finding the gene carriers among relatives and counseling [58,92]. In the absence of a family history, the different forms of acute porphyria may be distinguished by the characteristic patterns of porphyrin (and porphyrin-precursors) accumulation and excretion in plasma, urine, and stool. In case of increased urinary levels of ALA and PBG, fecal porphyrins should be measured: they are usually normal or minimally increased in AIP but elevated in HCP and VP [106]. It is worth reminding that some of these markers may be not present in the latent phase of the disease. Plasma fluorescence emission after excitation within the Soret band of light (at 410 nm of bandwidth) can be used to differentiate HCP and VP, which are characterized by different peak emissions [2,3,92,106,109]. Red blood cells activity of ALA-dehydratase and PBG-deaminase (Hydroxymethyl-bilane Synthase-HBMS) may be also readily assessed and can be helpful for establishing the diagnosis in ALAd-D porphyria and AIP, respectively [58]. Genetic testing may be helpful in the definitive diagnosis, especially when the type of disease and mutation are not already known from previous testing of relatives [58,110]. At present, accurate genotype-phenotype correlation for acute porphyrias is still ill-defined. Nevertheless, it is known the some types of gene mutations [such as Q180X, 198X and R173W mutations in PBGD (HBMS) gene, responsible for AIP, or R59W mutation in PPOX gene, responsible for VP] are associated with a significant higher penetrance of the diseases [52,53,111,112].
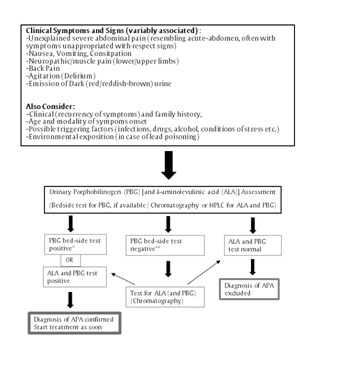 Figure 2:Schematic algorithm for diagnosis of Acute Porphyric Attack (APA).
Figure 2:Schematic algorithm for diagnosis of Acute Porphyric Attack (APA).
*Considering the extreme rarity of ALA dehydratase-deficiency porphyria (ALAD-P, which is characterized mostly by a greater ALA increase), the positivity of the “bedside” PBG assay, together with compatible clinical features, is highly suggestive of diagnosis of APA.
In case of “bedside” PBG test negative, the laboratory assessments for ALA and PBG may be relevant to rule out conditions characterized only by ALA increase (lead poisoning, ALAD-P or tyrosinemia).
**Check for ALAD-P; rule out tyrosinemia and lead poisoning.
THE ACUTE PORPHYRIC ATTACK: THE TREATMENT
The treatment of APA is the same apart from the kind of underlying acute porphyria (Table 1). Identification and elimination of all possible trigger factors (ex: drugs, infections and alcohol abuse) are mandatory. Except in the case of mild attacks, most patients require hospitalization and a continuous monitoring of neurologic status, blood pressure, heart rate, fluid and electrolyte balance, muscle, tendon and respiratory function. Symptoms (ex: systemic arterial hypertension, vomiting, seizures) should be treated avoiding porphyrinogenic drugs [7,25,70,92]. Opiates should be considered as mandatory in pain management during APA, being careful to prevent the opioid dependence in case of prolonged use [3,25].
Glucose down-regulates liver ALA-S (ALA-S1) (and hence ALA and PBG accumulation) and may help in resolving the crisis [113,114]. It can be administered by mouth in absence of nausea or vomiting, but, in most cases, it is given intravenously: the suggested regimen is 2-3 L of 10%-20% glucose (or dextrose) solution, infused by a central venous catheter over 24 h (100-125 mL/h). During the infusion period, it is important to carefully monitor the patient’s blood parameters, in order to avoid over hydration and/or electrolyte imbalance (especially hyponatremia) [3,25,92,115].
Heme infusion is much more effective than glucose in down-regulating ALA-S1 and should be used immediately in case of severe painful attacks, electrolyte imbalance or neurological involvement. It has been observed that in case of significant neuropathic involvement, a delay in heme infusion may be responsible for more severe nerve damage and a slower and possibly incomplete recovery of neuropathy [25].
In most cases, heme infusion resolves the APAs in 2-4 days. In U.S. heme is available as lyophilized hematin (Panhematin®), to be reconstituted with sterile water [116]. In Europe, heme is available as heme arginate (Normosang®) solution, to be diluted in 5% glucose or saline. In both cases, the recommended dose is 3 to 4 mg/kg IV once/day for 3-4 days [25,92,115-118]. Both hematin and heme arginate infusion may cause venous thrombosis and/or thrombophlebitis. Risk of these adverse events appears to be reduced if the heme is administered together with human serum albumin. Such binding also decreases the rate of development of hematin aggregates and improve the heme delivering to the tissues. For this reason, most experts recommend the infusion of heme (hematin or heme arginate) in association with human serum albumin [25,115]. In patients affected by severe forms of acute porphyrias (severe and recurrent attacks), who are considered at higher risk of kidney impairment or permanent neurologic damage, liver transplantation may be an option. Successful liver transplantation leads to permanent cure of all acute porphyrias. Patients with acute porphyrias should not serve as liver donors even though their liver may appear structurally normal (i.e., no cirrhosis), because recipients have developed APAs; such an outcome helped to confirm that the acute porphyrias are hepatic disorders [87,88,119-121]. Renal transplantation, with or without simultaneous liver exchange, should be considered in patients with active disease and terminal renal failure, because there is considerable risk that nerve damage will progress at the start of dialysis. The good results of recent experimental trials, based on gene-therapy approaches, depict new promising opportunities for the treatment of acute porphyrias, even in acute conditions [122-124].
THE ACUTE PORPHYRIC ATTACK: THE PREVENTION
All the people identified as carriers of an acute porphyria should carry a card attesting the carrier state and the precautions to be observed. The role of drugs in precipitating porphyric attacks, due to their possible effect on heme metabolism, is well-established; any drug treatment for a patient with porphyria should be prescribed according to an accurate reference to a drug list [3,20,70,125]. This is particularly important in surgery, many anesthetics being well-known powerful precipitant factor of acute attack [1,13,70,126,127]. Complete lists of potentially safe and unsafe drugs are continuously upgraded, and they are available on the net, at http://www.drugs-porphyria.org/ and http://www.porphyria-europe.com [89,128]. Some drugs are strictly forbidden, due to their well-depicted association with great number of severe attacks, however, most drugs are considered only potentially dangerous, and the majority of patients often tolerate them. For this last group of drugs, a common sense assessment of benefit versus risk is needed; an acute attack is in fact less likely to be precipitated in case of latent disease, or if the patient has experienced only a single attack, or if the concentrations of urinary metabolites (PBG, and particularly ALA) are within the normal range at the time of the prescription [7,70]. Alcohol abuse should be strictly forbidden, due its well-known effect in inducing heme metabolism. Due to the effect of glucose on heme metabolism, prolonged periods of starvation or drastic diets should be avoided: on the contrary a high-carbohydrate diet may decrease the risk and the frequency of acute attacks [3,7].
Patients who experience recurrent and predictable attacks (typically, women with attacks related to the menstrual cycle) may benefit from prophylactic heme therapy given shortly before the expected onset. No maintenance-standardized regimen is available: a specialist should be consulted [3,25,115]. Frequent premenstrual attacks in some women are aborted by administration of a gonadotropin-releasing hormone agonist plus low-dose estrogen. Low-dose oral contraceptives are sometimes used successfully, but the progestin component is likely to exacerbate the porphyria [129].
To prevent renal damage, chronic systemic arterial hypertension should be treated aggressively (using safe drugs). Patients with evidence of impaired renal function should be referred to a nephrologist. Due the defined higher risk for hepatocellular carcinoma, patients affected by acute porphyria should undergo twice-yearly surveillance, including liver screening with ultrasonography [7,78,130].
CONCLUSION
The acute porphyrias are rare diseases but their clinical exacerbations (acute porphyric attacks) may be potentially lethal conditions, especially if misdiagnosed or inappropriately treated.
The clinical features alone are not so specific and suitable to confirm a diagnosis of APA: for this reason, the knowledge and the correct interpretation of the appropriate tests are mandatory for an accurate clinical management of these diseases. In last decades, the availability of infusion-stable heme preparations has significantly improved the treatment outcome of APAs: for this reason, the knowledge about the diagnosis and the management of these diseases is relevant, especially for physicians working in internal and emergency medicine units.
CONFLICT-OF-INTEREST STATEMENT
Paolo Ventura has been involved as consultant in advisory boards and he has received research and travel grants from Orphan Europe Italy. There are no other competing interests to declare for him and for the other co-authors.
REFERENCES
- Puy H, Gouya L, Deybach JC (2010) Porphyrias. Lancet 375: 924-937.
- Kauppinen R (2005) Porphyrias. Lancet 365: 241-252.
- Ventura P (2011) Le Porfirie. In: Brunetti P, Santeusanio F, (eds). Trattato di Medicina Interna. Volume Malattie delle Ghiandole Endocrine, del Metabolismo e della Nutrizione. Piccin Nuova Libraria, Padova, Italy. Pg: 931-952.
- Desnick RJ (2002) Porfirie. In: Braunwald E, Fauci AS, Kasper DL, Hauser SL, Longo DL, Jameson JL, (eds). Harrison's: Principi di Medicina Interna. (15th edn) Milan: McGraw-Hill, USA. Pg: 2261-2302.
- Ventura E, Rocchi E (2001) Le Porfirie. In: Guarini G, Fiorelli G, Malliani A, Violi E, Volpe M, (eds). Teodori 2000 Trattato di Medicina Interna. (Volume 2. 6th edn) Roma: Società Editrice Universo, Italy. Pg: 2301-2334.
- Elder GH, Hift RJ, Meissner PN (1997) The acute porphyrias. Lancet 349: 1613-1617.
- Ventura P, Cappellini MD, Rocchi E (2009) The acute porphyrias: a diagnostic and therapeutic challenge in internal and emergency medicine. Intern Emerg Med 4: 297-308.
- Menegueti MG, Gil Cezar AT, Casarini KA, Muniz Cordeiro KS, Basile-Filho A, et al. (2011) Acute intermittent porphyria associated with respiratory failure: a multidisciplinary approach. Crit Care Res Pract 2011: 283690.
- Bylesjö I, Wikberg A, Andersson C (2009) Clinical aspects of acute intermittent porphyria in northern Sweden: a population-based study. Scand J Clin Lab Invest 69: 612-618.
- Solinas C, Vajda FJ (2008) Neurological complications of porphyria. J Clin Neurosci 15: 263-268.
- Pischik E, Kauppinen R (2009) Neurological manifestations of acute intermittent porphyria. Cell Mol Biol (Noisy-le-grand) 55: 72-83.
- Thunell S (2000) Porphyrins, porphyrin metabolism and porphyrias. I. Update. Scand J Clin Lab Invest 60: 509-540.
- James MF, Hift RJ (2000) Porphyrias. Br J Anaesth 85: 143-153.
- Gross U, Hoffmann GF, Doss MO (2000) Erythropoietic and hepatic porphyrias. J Inherit Metab Dis 23: 641-661.
- Chemmanur AT, Bonkovsky HL (2004) Hepatic porphyrias: diagnosis and management. Clin Liver Dis 8: 807-838, viii.
- Dombeck TA, Satonik RC (2005) The porphyrias. Emerg Med Clin North Am 23: 885-899, x.
- Poblete-Gutiérrez P, Wiederholt T, Merk HF, Frank J (2006) The porphyrias: clinical presentation, diagnosis and treatment. Eur J Dermatol 16: 230-240.
- Canavese C, Gabrielli D, Guida C, Cappellini MD (2002) [Nephrologists and porphyrias]. G Ital Nefrol 19: 393-412.
- Thunell S, Pomp E, Brun A (2007) Guide to drug porphyrogenicity prediction and drug prescription in the acute porphyrias. Br J Clin Pharmacol 64: 668-679.
- Gorchein A (1997) Drug treatment in acute porphyria. Br J Clin Pharmacol 44: 427-434.
- Deybach JC (2001) Porphyria. Painful photosensitivity. Lancet 358: 49.
- Stein PE, Badminton MN, Barth JH, Rees DC, Sarkany R, et al. (2012) Acute intermittent porphyria: fatal complications of treatment. Clin Med 12: 293-294.
- Kuo HC, Huang CC, Chu CC, Lee MJ, Chuang WL, et al. (2011) Neurological complications of acute intermittent porphyria. Eur Neurol 66: 247-252.
- Asselbergs FW, Kremer Hovinga TK, Bouwsma C, van Ingen J (2009) Acute intermittent porphyria as a cause of respiratory failure: case report. Am J Crit Care 18: 180, 178-179.
- Elder GH, Hift RJ (2001) Treatment of acute porphyria. Hosp Med 62: 422-425.
- Herrick AL, McColl KE, Moore MR, Cook A, Goldberg A (1989) Controlled trial of haem arginate in acute hepatic porphyria. Lancet 1: 1295-1297.
- Herrick A, McColl KE, McLellan A, Moore MR, Brodie MJ, et al. (1987) Effect of haem arginate therapy on porphyrin metabolism and mixed function oxygenase activity in acute hepatic porphyria. Lancet 2: 1178-1179.
- Anderson KE, Sassa S, Kappas A (1981) Acute intermittent porphyria. Ann Intern Med 95: 784-785.
- Grandchamp B (1998) Acute intermittent porphyria. Semin Liver Dis 18: 17-24.
- Anderson KE, Sassa S, Bishop DF, Desnick RJ (2001) Disorders of heme biosynthesis: X-linked sideroblastic anemia and the porphyrias. In: Scriver CR, Beaudet A, Sly WS, Valle D, (eds). The online metabolic and molecular basis of inherited diseases. McGraw-Hill, New York, USA. Pg: 2991-3062.
- Hift RJ, Peters TJ, Meissner PN (2012) A review of the clinical presentation, natural history and inheritance of variegate porphyria: its implausibility as the source of the 'Royal Malady'. J Clin Pathol 65: 200-205.
- Kirsch RE, Meissner PN, Hift RJ (1998) Variegate porphyria. Semin Liver Dis 18: 33-41.
- Di Pierro E, Ventura P, Brancaleoni V, Moriondo V, Marchini S, et al. (2009) Clinical, biochemical and genetic characteristics of Variegate Porphyria in Italy. Cell Mol Biol (Noisy-le-grand) 55: 79-88.
- Hift RJ, Meissner PN (2005) An analysis of 112 acute porphyric attacks in Cape Town, South Africa: Evidence that acute intermittent porphyria and variegate porphyria differ in susceptibility and severity. Medicine (Baltimore) 84: 48-60.
- Martásek P (1998) Hereditary coproporphyria. Semin Liver Dis 18: 25-32.
- Kühnel A, Gross U, Doss MO (2000) Hereditary coproporphyria in Germany: clinical-biochemical studies in 53 patients. Clin Biochem 33: 465-473.
- Sassa S (1998) ALAD porphyria. Semin Liver Dis 18: 95-101.
- Warren MJ, Cooper JB, Wood SP, Shoolingin-Jordan PM (1998) Lead poisoning, haem synthesis and 5-aminolaevulinic acid dehydratase. Trends Biochem Sci 23: 217-221.
- Herman DS, Geraldine M, Venkatesh T (2007) Evaluation, diagnosis, and treatment of lead poisoning in a patient with occupational lead exposure: a case presentation. J Occup Med Toxicol 2: 7.
- Bonkovsky HL, Guo JT, Hou W, Li T, Narang T, et al. (2013) Porphyrin and heme metabolism and the porphyrias. Compr Physiol 3: 365-401.
- Chen B, Hakenberg J, Sriniviasan R, Doheny DO, Peter I, et al. (2014) Acute intermittent porphyria: high incidence of pathogenic HMB-Synthase (HMBS) non-synonymous SNPs (nsSNPs) in genomic databases suggests other genetic/Environmental factors cause the acute attacks. Hepatology 60: 426A-4256B.
- Elder G, Harper P, Badminton M, Sandberg S, Deybach JC (2013) The incidence of inherited porphyrias in Europe. J Inherit Metab Dis 36: 849-857.
- Mustajoki P (1980) Variegate porphyria. Twelve years' experience in Finland. Q J Med 49: 191-203.
- Meissner PN, Meissner DM, Sturrock ED, Davidson B, Kirsch RE (1987) Porphyria--the UCT experience. S Afr Med J 72: 755-761.
- Nordmann Y, Puy H, Da Silva V, Simonin S, Robreau AM, et al. (1997) Acute intermittent porphyria: prevalence of mutations in the porphobilinogen deaminase gene in blood donors in France. J Intern Med 242: 213-217.
- Floderus Y, Shoolingin-Jordan PM, Harper P (2002) Acute intermittent porphyria in Sweden. Molecular, functional and clinical consequences of some new mutations found in the porphobilinogen deaminase gene. Clin Genet 62: 288-297.
- von und zu Fraunberg M, Pischik E, Udd L, Kauppinen R (2005) Clinical and biochemical characteristics and genotype-phenotype correlation in 143 Finnish and Russian patients with acute intermittent porphyria. Medicine (Baltimore) 84: 35-47.
- von und zu Fraunberg M, Timonen K, Mustajoki P, Kauppinen R (2002) Clinical and biochemical characteristics and genotype-phenotype correlation in Finnish variegate porphyria patients. Eur J Hum Genet 10: 649-657.
- Orphanet Report Series (2010) Prevalence of rare diseases: bibliographic data.
- Bonkovsky HL, Maddukuri VC, Yazici C, Anderson KE, Bissell DM, et al. (2014) Acute porphyrias in the USA: features of 108 subjects from Porphyrias Consortium. Am J Med 127: 1233-1241.
- Mustajoki P, Koskelo P (1976) Hereditary hepatic porphyrias in Finland. Acta Med Scand 200: 171-178.
- Meissner PN, Corrigall AV, Hift RJ (2012) Fifty years of porphyria at the University of Cape Town. S Afr Med J 102: 422-426.
- Meissner PN, Dailey TA, Hift RJ, Ziman M, Corrigall AV, et al. (1996) A R59W mutation in human protoporphyrinogen oxidase results in decreased enzyme activity and is prevalent in South Africans with variegate porphyria. Nat Genet 13: 95-97.
- Kauppinen R, Mustajoki P (1992) Prognosis of acute porphyria: occurrence of acute attacks, precipitating factors, and associated diseases. Medicine (Baltimore) 71: 1-13.
- Hift RJ, Meissner D, Meissner PN (2004) A systematic study of the clinical and biochemical expression of variegate porphyria in a large South African family. Br J Dermatol 151: 465-471.
- WALDENSTROM J, HAEGER-ARONSEN B (1963) Different patterns of human porphyria. Br Med J 2: 272-276.
- Jeans JB, Savik K, Gross CR, Weimer MK, Bossenmaier IC, et al. (1996) Mortality in patients with acute intermittent porphyria requiring hospitalization: a United States case series. Am J Med Genet 65: 269-273.
- Ventura P, Cappellini MD, Biolcati G, Guida CC, Rocchi E (2014) A challenging diagnosis for potential fatal diseases: recommendations for diagnosing acute porphyrias. Eur J Intern Med 25: 497-505.
- Bautista O, Vázquez-Caubet JC, Zhivago EA, Dolores Sáiz M (2014) From metabolism to psychiatric symptoms: psychosis as a manifestation of acute intermittent porphyria. J Neuropsychiatry Clin Neurosci 26: 30.
- Kumar B (2012) Acute intermittent porphyria presenting solely with psychosis: a case report and discussion. Psychosomatics 53: 494-498.
- Massey EW (1980) Neuropsychiatric manifestations of porphyria. J Clin Psychiatry 41: 208-213.
- Scott L, Bean DW (1987) Psychiatric manifestations of porphyria. S D J Med 40: 5-8.
- Crimlisk HL (1997) The little imitator--porphyria: a neuropsychiatric disorder. J Neurol Neurosurg Psychiatry 62: 319-328.
- Usalan C, Erdem Y, Altun B, Gürsoy M, Celik I, et al. (1996) Severe hyponatremia due to SIADH provoked by acute intermittent porphyria. Clin Nephrol 45: 418.
- Marsden JT, Rees DC (2014) Urinary excretion of porphyrins, porphobilinogen and δ-aminolaevulinic acid following an attack of acute intermittent porphyria. J Clin Pathol 67: 60-65.
- Hift RJ (1999) The diagnosis of porphyria. S Afr Med J 89: 611-614.
- Hultdin J, Schmauch A, Wikberg A, Dahlquist G, Andersson C (2003) Acute intermittent porphyria in childhood: a population-based study. Acta Paediatr 92: 562-568.
- Kauppinen R, von und zu Fraunberg M (2002) Molecular and biochemical studies of acute intermittent porphyria in 196 patients and their families. Clin Chem 48: 1891-1900.
- Zhao B, Wei Q, Wang Y, Chen Y, Shang H (2014) Posterior reversible encephalopathy syndrome in acute intermittent porphyria. Pediatr Neurol 51: 457-460.
- Roveri G, Nascimbeni F, Rocchi E, Ventura P (2014) Drugs and acute porphyrias: reasons for a hazardous relationship. Postgrad Med 126: 108-120.
- Bargetzi MJ, Meyer UA, Birkhaeuser MH (1989) Premenstrual exacerbations in hepatic porphyria: prevention by intermittent administration of an LH-RH agonist in combination with a gestagen. JAMA 261: 864.
- Aggarwal N, Bagga R, Sawhney H, Suri V, Vasishta K (2002) Pregnancy with acute intermittent porphyria: a case report and review of literature. J Obstet Gynaecol Res 28: 160-162.
- Kanaan C, Veille JC, Lakin M (1989) Pregnancy and acute intermittent porphyria. Obstet Gynecol Surv 44: 244-249.
- Andersson C, Lithner F (1994) Hypertension and renal disease in patients with acute intermittent porphyria. J Intern Med 236: 169-175.
- Coban-Karatas M, Erol I, Ozkale Y, Yazici N (2013) Central retinal artery occlusion in a 13-year-old child as a presenting sign of hyperhomocysteinemia together with high lipoprotein (a) level. Pediatr Neurol 49:138-140.
- Church SE, McColl KE, Moore MR, Youngs GR (1992) Hypertension and renal impairment as complications of acute porphyria. Nephrol Dial Transplant 7: 986-990.
- Andant C, Puy H, Bogard C, Faivre J, Soulé JC, et al. (2000) Hepatocellular carcinoma in patients with acute hepatic porphyria: frequency of occurrence and related factors. J Hepatol 32: 933-939.
- Schneider-Yin X, van Tuyll van Serooskerken AM, Went P, Tyblewski W, Poblete-Gutiérrez P, et al. (2010) Hepatocellular carcinoma in variegate porphyria: a serious complication. Acta Derm Venereol 90: 512-515.
- Stewart MF (2012) Review of hepatocellular cancer, hypertension and renal impairment as late complications of acute porphyria and recommendations for patient follow-up. J Clin Pathol 65: 976-980.
- Rocchi E, Ventura P, Ronzoni A, Rosa MC, Gozzi C, et al. (2004) Pro-oxidant and antioxidant factors in acute intermittent porphyria: family studies. J Inherit Metab Dis 27: 251-266.
- Lindberg RL, Martini R, Baumgartner M, Erne B, Borg J, et al. (1999) Motor neuropathy in porphobilinogen deaminase-deficient mice imitates the peripheral neuropathy of human acute porphyria. J Clin Invest 103: 1127-1134.
- Lindberg RL, Porcher C, Grandchamp B, Ledermann B, Bürki K, et al. (1996) Porphobilinogen deaminase deficiency in mice causes a neuropathy resembling that of human hepatic porphyria. Nat Genet 12: 195-199.
- Shanley BC, Neethling AC, Percy VA, Carstens M (1975) Neurochemical aspects of porphyria. Studies on the possible neurotoxicity of delta-aminolaevulinic acid. S Afr Med J 49: 576-580.
- Badawy AA (1978) Tryptophan pyrrolase, the regulatory free haem and hepatic porphyrias. Early depletion of haem by clinical and experimental exacerbators of porphyria. Biochem J 172: 487-494.
- Homedan C, Laafi J, Schmitt C, Gueguen N, Lefebvre T, et al. (2014) Acute intermittent porphyria causes hepatic mitochondrial energetic failure in a mouse model. Int J Biochem Cell Biol 51: 93-101.
- Meyer UA, Schuurmans MM, Lindberg RL (1998) Acute porphyrias: pathogenesis of neurological manifestations. Semin Liver Dis 18: 43-52.
- Ponka P (1997) Tissue-specific regulation of iron metabolism and heme synthesis: distinct control mechanisms in erythroid cells. Blood 89: 1-25.
- Seth AK, Badminton MN, Mirza D, Russell S, Elias E (2007) Liver transplantation for porphyria: who, when, and how? Liver Transpl 13: 1219-1227.
- Singal AK, Parker C, Bowden C, Thapar M, Liu L, et al. (2014) Liver transplantation in the management of porphyria. Hepatology 60: 1082-1089.
- European Porphyria Initiative
- Bonkovsky HL, Barnard GF (1998) Diagnosis of porphyric syndromes: a practical approach in the era of molecular biology. Semin Liver Dis 18: 57-65.
- de Rooij WM, Edixhoven A, Wilson JH (2003) Porphyria: a diagnostic approach. In: Kadish KM, Smith KM, Guillard R, (eds). The porphyrin handbook. Elsevier, St Louis,USA. Pg: 211-245.
- Anderson KE, Bloomer JR, Bonkovsky HL, Kushner JP, Pierach CA, et al. (2005) Recommendations for the diagnosis and treatment of the acute porphyrias. Ann Intern Med 142: 439-450.
- Tran TP, Leduc K, Savard M, Dupré N, Rivest D, et al. (2013) Acute porphyria presenting as epilepsia partialis continua. Case Rep Neurol 5: 116-124.
- Bottomley SS, Bonkowsky HL, Kreimer-Birnbaum M (1981) The diagnosis of acute intermittent porphyria. Usefulness and limitations of the erythrocyte uroporphyrinogen I synthase assay. Am J Clin Pathol 76: 133-139.
- Bissell DM (1982) Laboratory evaluation in porphyria. Semin Liver Dis 2: 100-107.
- Lamon J, With TK, Redeker AG (1974) The Hoesch test: bedside screening for urinary porphobilinogen in patients with suspected porphyria. Clin Chem 20: 1438-1440.
- Thunell S, Harper P, Brock A, Petersen NE (2000) Porphyrins, porphyrin metabolism and porphyrias. II. Diagnosis and monitoring in the acute porphyrias. Scand J Clin Lab Invest 60: 541-559.
- Andersson C, Thunell S, Floderus Y, Forsell C, Lundin G, et al. (1995) Diagnosis of acute intermittent porphyria in northern Sweden: an evaluation of mutation analysis and biochemical methods. J Intern Med 237: 301-308.
- Henderson MJ (1989) Thin-layer chromatography of free porphyrins for diagnosis of porphyria. Clin Chem 35: 1043-1044.
- Lai CK, Lam CW, Chan YW (1994) High-performance thin-layer chromatography of free porphyrins for diagnosis of porphyria. Clin Chem 40: 2026-2029.
- Sardh E, Andersson DE, Henrichson A, Harper P (2009) Porphyrin precursors and porphyrins in three patients with acute intermittent porphyria and end-stage renal disease under different therapy regimes. Cell Mol Biol (Noisy-le-grand) 55: 66-71.
- Sardh E, Harper P, Andersson DE, Floderus Y (2009) Plasma porphobilinogen as a sensitive biomarker to monitor the clinical and therapeutic course of acute intermittent porphyria attacks. Eur J Intern Med 20: 201-207.
- With TK (1971) Simple and rapid screening for acute porphyria--'porphobilistix' and Hoesch test. S Afr Med J 25: 229-230.
- Deacon AC, Peters TJ (1998) Identification of acute porphyria: evaluation of a commercial screening test for urinary porphobilinogen. Ann Clin Biochem 35 : 726-732.
- McEwen J, Paterson C (1972) Drugs and false-positive screening tests for porphyria. Br Med J 1: 421.
- Sandberg S, Elder GH (2004) Diagnosing acute porphyrias. Clin Chem 50: 803-805.
- Beard D (1993) Plumbo porphyria. A difficult diagnosis. Occup Health (Lond) 45: 25.
- Frith D, Yeung K, Thrush S, Hunt BJ, Hubbard JG (2005) Lead poisoning--a differential diagnosis for abdominal pain. Lancet 366: 2146.
- Walsh DS, Beard JS, James WD (1994) Fluorescent spectrophotometric analysis in the evaluation of porphyria. JAMA 272: 1580-1581.
- Whatley SD, Badminton MN (2013) Role of genetic testing in the management of patients with inherited porphyria and their families. Ann Clin Biochem 50: 204-216.
- Andersson C, Floderus Y, Wikberg A, Lithner F (2000) The W198X and R173W mutations in the porphobilinogen deaminase gene in acute intermittent porphyria have higher clinical penetrance than R167W. A population-based study. Scand J Clin Lab Invest 60: 643-648.
- Paradisi I, Arias S (2010) Marked geographic aggregation of acute intermittent porphyria families carrying mutation Q180X in Venezuelan populations, with description of further mutations. J Inherit Metab Dis 3: 455-463.
- Doss M, Verspohl F (1981) The "glucose effect" in acute hepatic porphyrias and in experimental porphyria. Klin Wochenschr 59: 727-735.
- Handschin C, Lin J, Rhee J, Peyer AK, Chin S, et al. (2005) Nutritional regulation of hepatic heme biosynthesis and porphyria through PGC-1alpha. Cell 122: 505-515.
- Stein P, Badminton M, Barth J, Rees D, Stewart MF; British and Irish Porphyria Network (2013) Best practice guidelines on clinical management of acute attacks of porphyria and their complications. Ann Clin Biochem 50: 217-223.
- Anderson KE, Bonkovsky HL, Bloomer JR, Shedlofsky SI (2006) Reconstitution of hematin for intravenous infusion. Ann Intern Med 144: 537-538.
- Loftin EB 3rd (1985) Hematin therapy in acute porphyria. JAMA 254: 613.
- Pierach CA (1989) Haem and porphyria attacks. See comment in PubMed Commons below Lancet 2: 213-214.
- Soonawalla ZF, Orug T, Badminton MN, Elder GH, Rhodes JM, et al. (2004) Liver transplantation as a cure for acute intermittent porphyria. Lancet 363: 705-706.
- Dar FS, Asai K, Haque AR, Cherian T, Rela M, et al. (2010) Liver transplantation for acute intermittent porphyria: a viable treatment? Hepatobiliary Pancreat Dis Int 9: 93-96.
- Dowman JK, Gunson BK, Bramhall S, Badminton MN, Newsome PN (2011) Liver transplantation from donors with acute intermittent porphyria. Ann Intern Med 154: 571-572.
- Yasuda M, Gan L, Chen B, Kadirvel S, Yu C, et al. (2014) RNAi-mediated silencing of hepatic Alas1 effectively prevents and treats the induced acute attacks in acute intermittent porphyria mice. Proc Natl Acad Sci USA
- [No authors listed] (2014) Augmenting PBGD expression in the liver as a novel gene therapy for acute intermittent porphyria (AIPgene). Hum Gene Ther Clin Dev 25: 61-63.
- Pañeda A, Lopez-Franco E, Kaeppel C, Unzu C, Gil-Royo AG, et al. (2013) Safety and liver transduction efficacy of rAAV5-cohPBGD in nonhuman primates: a potential therapy for acute intermittent porphyria. Hum Gene Ther 24: 1007-1017.
- Hift RJ, Thunell S, Brun A (2011) Drugs in porphyria: From observation to a modern algorithm-based system for the prediction of porphyrogenicity. Pharmacol Ther 132: 158-169.
- Ashley EM (1996) Anaesthesia for porphyria. Br J Hosp Med 56: 37-42.
- Baker SD, Taylor B (2000) Anaesthesia is also risky in patients with porphyria. BMJ 321: 1023.
- NAPOS. The Drug Database for Acute Porphyria.
- Anderson KE, Spitz IM, Bardin CW, Kappas A (1990) A gonadotropin releasing hormone analogue prevents cyclical attacks of porphyria. Arch Intern Med 150: 1469-1474.
- Sardh E, Wahlin S, Björnstedt M, Harper P, Andersson DE (2013) High risk of primary liver cancer in a cohort of 179 patients with Acute Hepatic Porphyria. J Inherit Metab Dis 36: 1063-1071.
Citation: Ventura P, Cuoghi C, Marcacci M (2015) The Acute Porphyric Attack: A Difficult Diagnosis for a Potential Lethal Event in Emergency Medicine. J Emerg Med Trauma Surg Care 2: 005.
Copyright: © 2015 Chiara Cuoghi, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.