Assessment of Four Polymerases for Low Volume, Fast PCR Amplification of STR Loci for DNA Reference Samples
*Corresponding Author(s):
Catherine C ConnonDepartment Of Forensic Science, Cellmark Forensics, A LabCorp Specialty Testing Group, Department Of Biological Sciences, University Of North Texas, Virginia Commonwealth University, Richmond, Virginia, Texas, United States
Tel:+1 8048284318,
Fax:+1 8048284983
Email:cmconnon@vcu.edu
Abstract
The goal of this project was to evaluate several commercially available products for their suitability with fast PCR amplification of forensic STR loci using a previously validated low volume reaction (3μl) in a non-fast thermal cycler for single-source reference DNA samples. This project was the first step associated with developing multiple low volume (3-6µl), fast PCR reactions for several primer sets commonly used in the forensic community with the ultimate goal of reducing amplification time and cost, while still producing STR profiles of sufficient quality from single-source reference samples. Four products (AmpliTaq Gold® Fast PCR Master Mix, KAPA2G™ Fast Multiplex PCR Kit, SpeedSTAR™ HS DNA Polymerase and Type-it Microsatellite PCR Kit) were evaluated and optimized for low volume (3μl) fast PCR on a 384-well Veriti® thermal cycler with the AmpF?STR® Identifiler® PCR Amplification Kitprimer set, and resulted in amplification times of 43 min (KAPA2G) to 1 hr 19 min (AmpliTaq Gold Fast). Two-step PCR cycling utilizing a combined annealing/extension step was successful with KAPA2G and Type-it fast PCR protocols, but not AmpliTaq Gold Fast or Speed STAR. Once optimized protocols were established for each product, several comparison studies were conducted, including: 1) determination of optimal DNA input ranges (profiles were evaluated via sensitivity, reproducibility of peak height, inter- and intra-locus peak balance, stutter, pull-up, -A, specificity and background noise); 2) determination of stochastic thresholds; 3) precision; 4) stutter assessment; and 5) optimization checks to determine compatibility with automation and overall profile “pass” rate. In addition to profile quality, amplification time, reagent cost, and ease of PCR setup were also taken into account. Using all of these criteria, KAPA2G™ Fast Multiplex PCR Kit was selected as the prime candidate for future studies involving development of low volume, fast PCR protocols for several primer sets commonly used in the forensic community, as it demonstrated ~2 hr reduction (73%) in amplification time and a 92% first pass success rate for buccal swab samples.
Keywords
INTRODUCTION
Fast PCR and/or direct PCR are fairly recent improvements from the late 2000s/early 2010s that can be utilized by the forensic DNA community to reduce processing time and/or costs for forensic-type and reference samples [1-10]. Reduced volume PCR amplification has been shown to improve sensitivity and efficiency [11], but the more reduced the total reaction volume becomes, the more intra-locus peak balance decreases, making extremely low volume reactions likely unsuitable for anything other than single-source reference samples. Cellmark Forensics successfully utilized low volume PCR reactions of 3-6µl for numerous primer sets commonly used within the forensic community in their databasing unit from 2008-2015. These low volume reactions were coupled with a PicoGreen® quantification detection system, and though not-human specific, this was acceptable by the FBI Quality Assurance Standards guidelines for reference samples in both casework and databasing laboratories because the system had been validated and proven to be reproducible and reliable [12, 13]. Using a PicoGreen® quantification method followed by low volume amplification, Cellmark’s databasing unit achieved a first pass success of >90% for multiple primer sets and reference sample types (including buccal samples, which also contain bacterial DNA that contributes to a portion of the quantification value obtained) during the 7-year duration in which the above mentioned work flow was utilized.
In 2008, Vallone et al., demonstrated that fast PCR could be achieved using a non-fast thermal cycler - the GeneAmp® PCR System 9700 (9700; Applied Biosystems, Foster City, CA) - and the AmpF?STR® Identifiler® PCR Amplification Kit (Identifiler; Applied Biosystems) primer set with a 10μl PCR reaction in less than 36 min [8]. Using fast thermal cyclers, fast PCR has been achieved in as little as 19-26 min [1,2,5]. In general, fast PCR profiles have often suffered from low level artifacts, increased n-4 stutter percentage and incomplete Adenylation (-A) [1,2,5,8-10].
More recently, rapid DNA testing has been achieved in less than two hours from start to finish using a single instrument, and has demonstrated its suitability for investigative leads when time is a crucial factor [14,15], though it is not a cost effective means of testing for high-throughput laboratories. For high-throughput laboratories, time and cost effectiveness are critical, and often, the means to achieving a reduction in processing time includes added costs (e.g., use of fast thermal cyclers and fast polymerases).
This paper focuses on achieving fast PCR for low volume reactions (e.g., 3µl) without increasing costs, while maintaining high quality STR profiles. A standard, non-fast thermal cycler was utilized for PCR, rather than more expensive fast thermal cycler alternatives. Additionally, to offset added reagent costs associated with utilizing fast polymerases, extremely reduced amplification volumes (3μl) were assessed, which would not be suitable for direct amplification. Four commercially available products were assessed for fast PCR suitability with the Identifiler primer set using a 3μl reaction carried out on a 384-well Veriti® (Veriti; Applied Biosystems) thermal cycler: AmpliTaq Gold® Fast PCR Master Mix (AmpliTaq Gold Fast; Applied Biosystems), KAPA2G™ Fast Multiplex PCR Kit (KAPA2G; Kapa Biosystems Inc., Woburn, MA), Speed STAR™ HS DNA Polymerase (Speed STAR; Takara Bio Inc., Shiga, Japan) and Type-it Microsatellite PCR Kit (Type-it; QIAGEN, Valencia, CA). Selection of these products was based on a variety of criteria, including low cost, being a non-template adenylator, and discussions with manufacturers to identify which of their products had a high potential to perform fast PCR multiplexing, as well as some previous studies on fast PCR [1,2,5,8]. All of these products are (or contain) fast polymerases, but Type-it was not specifically designed for fast PCR. The Identifiler primer set consists of 15 STR loci and a sex marker (Amelogenin), therefore the products chosen would also have to be able to perform under multiplexing conditions. However, only KAPA2G and Type-it were specifically designed for multiplexing. Thus, of the products chosen for evaluation, one was developed for fast PCR and multiplexing (KAPA2G), two for fast PCR but not multiplexing (AmpliTaq Gold Fast and SpeedSTAR), and one for multiplexing but not fast PCR (Type-it, although it contains HotStarTaq® Plus DNA polymerase, which is a fast polymerase).
The prospect of coupling fast PCR with low volume reactions would be of tremendous benefit for the processing of single-source DNA reference samples via a significant decrease in processing time, thereby allowing a substantial increase in sample throughput. Furthermore, the goal was to accomplish these process improvements without incurring additional reagent or equipment costs and while maintaining high STR profile quality. Additionally, a substantial cost savings would be realized for those laboratories that are currently using full or half reactions for their reference samples.
MATERIALS AND METHODS
This study began with the optimization of low volume (3µl), fast PCR protocols for each of the four products evaluated - AmpliTaq Gold Fast, KAPA2G, SpeedSTAR, and Type-it - with the Identifiler primer set to amplify buccal swab samples on a Veriti (non-fast) thermal cycler. Following optimization of each product, a thorough comparison study was conducted between these four protocols and that of the previously validated 3µl “standard” (i.e., non-fast PCR) Identifiler amplification protocol utilized by Cellmark Forensics to determine which low volume, fast PCR protocol (if any) offered process improvements via a decrease in amplification time without compromising STR profile quality. It should be noted that the initial optimization of each product was based on amplification of DNA from two individuals (processed in triplicate), and then the comparison study included testing DNA from an additional 25 individuals. These studies are discussed in more detail below.
For all studies, buccal swab cuttings (~¼ swab) were obtained from a total of 27 individuals and were extracted (quarter reaction with a minimum of a one hour incubation at 56°C) using the ChargeSwitch® Forensic DNA Purification Kit (ChargeSwitch; Applied Biosystems) [16] on a BioSprint 96 (QIAGEN) or KingFisher® 96 (Thermo Scientific, Vantaa, Finland). Samples were then quantified using the Quant-iT™ PicoGreen® dsDNA Quantitation Kit coupled with the Quant-iT™ PicoGreen® dsDNA Quantitation Reagent (PicoGreen; Applied Biosystems) and a FLUO star microplate reader (BMG LABTECH, Ortenberg, Germany), followed by a pre-amplification dilution to normalize samples for amplification. Amplification is discussed in more detail below (Table 1 includes reaction composition). Following amplification, amplification product was diluted by the addition of 4μl sterile water to the entire 3µl amplification product reactions. One microliter of diluted amplification product was combined with 10μl of a formamide/size standard mixture (10μl formamide and 0.2μl GeneScan™ 500 LIZ™ [LIZ; Applied Biosystems]) for each sample or allelic ladder. Prepared amplification product was detected using a 3130xl Genetic Analyzer (3130xl; Applied Biosystems) equipped with POP-4® (POP-4; Applied Biosystems) and a 36cm array using a 3kV, 7sec injection. All profiles were analyzed with GeneMapper™ ID v3.2 software using a 75rfu threshold. Specific analysis is discussed below. Any tests for statistical significance were performed using a 5% significance level.
| AmpliTaq Gold Fast | KAPA2G | SpeedSTAR | Type-it | Standard Identifiler |
| 1.5µl AmpliTaq Gold® Fast PCR Master Mix (2X) | 1.5µl KAPA2G™ Fast Multiplex Mix | 0.3µl 10X Fast Buffer I | 1.5µl Type-it Multiplex PCR Master Mix | 1.145µlPCR Reaction Mix |
| 0.6µl Identifiler Primers | 0.24µl dNTPs | 0.055µl AmpliTaq Gold | ||
| 0.9µl DNA Template | 0.6µl Identifiler Primers | 0.015µl Speed STAR™ HS | 0.6µl Identifiler Primers | |
| 0.9µl DNA Template | 0.6µl Identifiler Primers | 0.9µl DNA Template | 0.6µl Identifiler Primers | |
| 0.9µl DNA Template | 0.9µl DNA Template | |||
| 0.945µl Water | 0.3µl Water |
OPTIMIZATION OF LOW VOLUME, FAST PCR PROTOCOLS
AmpliTaq Gold® fast PCR master mix
| PCR Step | AmpliTaq Gold Fast | KAPA2G | SpeedSTAR | Type-it | Standard Identifiler |
| Polymerase Activation | 95°C 10min | 95°C 1min | 95°C 1min | 95°C 5min | 95°C 11min |
| 26 Cycles of: | |||||
| Denaturation | 96°C 10sec | 95°C 5sec | 98°C 5sec | 96°C 30sec | 94°C 1min |
| Annealing | 61°C 45sec | 61°C 40sec | 61°C 25sec | 59°C 1min 15sec | 59°C 1min |
| Extension | 68°C 45sec | 72°C 20sec | 72°C 1min | ||
| Final Extension | 72°C 13min | 72°C 10min | 72°C 13min | 72°C 10min | 60°C 60min |
| Hold | 25°C | 25°C | 25°C | 25°C | 4°C |
| Total Time | 1hr 19min | 43min | 49min | 1hr 14min | 2hr 42min |
| Time Saved | 1hr 23min | 1hr 59min | 1hr 53min | 1hr 28min | |
| -51% | -73% | -70% | -54% | ||
| Added Cost | $0.10 | $0.06 | $0.06 | $0.13 |
| 3-Step PCR | 2-Step PCR | |||||||
| PCR Step | Final Extension | Primer Annealing | Denaturation & Annealing/Extension | Denaturation | Initial Activation | Final Hold | Annealing/ Extension | Denaturation |
| Polymerase Activation | 95°C 10min | 95°C 10min | 95°C 10min | 95°C 10min | 95°C 1, 3, 10min | 95°C 10min | 95°C 10min | 95°C 10min |
| 26 Cycles of: | ||||||||
| Denaturation | 96°C 5sec | 96°C 5sec | 96°C 5, 10sec | 96°C 15, 20sec | 96°C 10sec | 96°C 10sec | 96°C 10sec | 96°C 10sec, 1min |
| Annealing | 59°C 10sec | 59, 61, 63°C 50sec | 61°C 40, 50, 60sec | 61°C 1min | 61°C 1min | 61°C 1min | 61°C 1, 2min | 61°C 2min |
| Extension | 68°C 10sec | |||||||
| Final Extension | 72°C 1, 5, 10min | 72°C 10min | 72°C 10min | 72°C 10min | 72°C 10min | 72°C 10min | 72°C 10min | 72°C 10min |
| Hold | 4°C | 4°C | 4°C | 4°C | 4°C | 4, 25°C | 25°C | 25°C |
| Total Time | 35, 39, 44min | 58, 57, 56min | 52min, 54min, 56min, 58min, | 1hr 5min, | 54min, 55min, | 1hr 3min | 1hr 29min | 1hr 29min, |
| 1hr 7min | 1hr 51min | |||||||
| 1hr 1min, 1hr 3min | 1hr 3min | |||||||
| 3-Step PCR | ||||||||
| PCR Step | Standard PCR | Annealing & Extension | Denaturation | Annealing | Extension | Final Extension | Final | |
| Polymerase Activation | 95°C 11min | 95°C 10min | 95°C 10min | 95°C 10min | 95°C 10min | 95°C 10min | 95°C 10min | |
| 26 Cycles of: | ||||||||
| Denaturation | 94°C 1min | 96°C 1min | 96°C 10sec, 1min | 96°C 10sec | 96°C 10sec | 96°C 10sec | 96°C 10sec | |
| Annealing | 59°C 1min | 61°C 1min | 61°C 1min | 61°C 15, 30, 45, 60sec | 61°C 45sec | 61°C 45sec | 61°C 45sec | |
| Extension | 72°C 1min | 68, 72°C 1min | 68°C 1min | 68°C 1min | 68°C 30, 45, 60sec | 68°C 45sec | 68°C 45sec | |
| Final Extension | 60°C 60min | 72°C 10min | 72°C 10min | 72°C 10min | 72°C 10min | 72°C 10, 13min | 72°C 13min | |
| Hold | 4°C | 25°C | 25°C | 25°C | 25°C | 25°C | 25°C | |
| Total Time | 2hr 42min | 1hr 51min | 1hr 29min | 1hr 10min, 1hr 16min, 1hr 23min, 1hr 29min | 1hr 10min, 1hr 16min, 1hr 23min | 1hr 16min, | 1hr 19min | |
| 1hr 19min | ||||||||
The PCR step under evaluation is listed at the top of each column. Each of the thermal cycling parameters was assessed using a 384-well Veriti thermal cycler. PHR <50% were problematic and resulted testing of more parameters.
KAPA2G™ fast multiplex PCR kit
The PCR step under evaluation is listed at the top of each column. Each of the thermal cycling parameters was assessed using a 384-well Veriti thermal cycler.
| 3-Step PCR | 2-Step PCR | ||||||
| PCR Step | Final Extension | Primer Annealing | Denaturation | Annealing/Extension | Initial Activation | Final Extension | Final |
| Polymerase Activation | 95°C 3min | 95°C 3min | 95°C 3min | 95°C 3min | 95°C 1, 2, 3min | 95°C 1min | 95°C 1min |
| 26 Cycles of: | |||||||
| Denaturation | 95°C 15sec | 95°C 15sec | 95°C 5, 10, 15sec | 95°C 5sec | 95°C 5sec | 95°C 5sec | 95°C 5sec |
| Annealing | 59°C 30sec | 59, 61, 63°C 30sec | 61°C 30sec | 61°C 30, 35, 40sec | 61°C 40sec | 61°C 40sec | 61°C 40sec |
| Extension | 72°C 30sec | ||||||
| Final Extension | 72°C 1, 2, 5min | 72°C 5min | 72°C 5min | 72°C 5min | 72°C 5min | 72°C 3, 4, 5, 10min | 72°C 10min |
| Hold | 25°C | 25°C | 25°C | 25°C | 25°C | 25°C | 25°C |
| Total Time | 50, 51, 54min | 41, 40, 39min | 36, 38, 40min | 36, 38, 40min | 38, 39, 40min | 36, 37, 38, 43min | 43min |
Table 4: Thermal Cycling Parameters for 3µl Identifiler Fast PCR Development with KAPA2G.
SpeedSTAR™ HS DNA polymerase
| 2-Step PCR | ||||||
| PCR Step | Primer Annealing | Annealing/Extension | Denaturation & Annealing/Extension | |||
| Polymerase Activation | 95°C 1min | 95°C 1min | 95°C 1min | |||
|
26 Cycles of: |
95°C 5sec |
95°C 5sec |
95°C 5, 10sec |
|||
| Final Extension | 72°C 1min | 72°C 1min | 72°C 1min | |||
| Hold | 25°C | 25°C | 25°C | |||
| Total Time | 28, 27mina | 28, 29, 38min | 28-49min | |||
| 3-Step PCR | ||||||
| PCR Step | Primer Annealing | Extension | Annealing | Final Extension | Primer Annealing | Final |
| Polymerase Activation | 95°C 1min | 95°C 1min | 95°C 1min | 95°C 1min | 95°C 1min | 95°C 1min |
|
26 Cycles of: |
98°C 5sec |
98°C 5sec |
98°C 5sec |
98°C 5sec |
98°C 5sec |
98°C 5sec |
| Final Extension | 72°C 1min | 72°C 1min | 72°C 1min | 72°C 1, 10, 13min | 72°C 13min | 72°C 13min |
| Hold | 25°C | 25°C | 25°C | 25°C | 25°C | 25°C |
| Total Time | 29, 28mina | 29, 33min | 33, 38min | 38, 47, 50min | 50, 49, 48min | 49min |
Table 5: Thermal Cycling Parameters for 3µl Identifiler Fast PCR Development with SpeedSTAR.
The PCR step under evaluation is listed at the top of each column. Each of the thermal cycling parameters was assessed using a 384-well Veriti thermal cycler. For 2-step PCR cycles, the manufacturer recommends a denaturation temperature of 95°C, but 98°C for 3-step cycling.
aFor 61°C and 63°C protocols.
Type-it® microsatellite PCR kit
The PCR step under evaluation is listed at the top of each column. Each of the thermal cycling parameters was assessed using a 384-well Veriti thermal cycler.
| 3-Step PCR | 2-Step PCR | |||||
| PCR Step | Longer Cycles | Longer Cycles | Initial Activation | Denaturation | Annealing/ Extension | Final |
| Polymerase Activation | 95°C 5min | 95°C 5min | 95°C 1, 2, 5min | 95°C 5min | 95°C 5min | 95°C 5min |
|
26 Cycles of: |
95°C 30sec |
95°C 30sec |
95°C 30sec |
95°C 10, 20, 30sec |
95°C 30sec 59°C 55sec, 1min 5sec, 1min 15sec |
96°C 30sec |
| Final Extension | 72°C 10min | 72°C 10min | 72°C 10min | 72°C 10min | 72°C 10min | 72°C 10min |
| Hold | 25°C | 25°C | 25°C | 25°C | 25°C | 25°C |
| Total Time | 1hr 13min | 1hr 14min | 1hr 9min, 1hr 10min, 1hr 14min | 1hr 5min, 1hr 9min, 1hr 14min | 1hr 5min, 1hr 9min, 1hr 14min | 1hr 14min |
Table 6: Thermal Cycling Parameters for 3µl Identifiler Fast PCR Development with Type-it.
Comparison of Four Low Volume, Fast PCR Protocols to Low Volume, Standard PCR
The four fast PCR protocols were compared to each other and to standard Identifiler amplification with regard to amplification time, fast PCR reagent cost and overall performance. PCR performance was evaluated based on optimal DNA input ranges (determined by sensitivity, reproducibility, inter-locus balance, intra-locus balance, stutter, pull-up, -A, non-specific amplification and baseline noise), stochastic threshold, precision of allele sizing, stutter and an optimization check consisting of 25 samples. Each of these analyses is discussed in more detail below.
Determination of the optimal DNA input ranges
Sensitivity: Sensitivity range was a key component for the determination of DNA input range; this was defined as the lowest DNA input amount from which full profiles were obtained from the majority of the samples, through the highest DNA input from which full profiles were obtained from the majority of the samples, using a 75rfu threshold. Full profiles had to be obtained at each of the DNA inputs throughout the entire sensitivity range. Unless otherwise noted, all additional analyses for the determination of the optimal DNA input range only utilized data from within the sensitivity range.
Reproducibility: Reproducibility of allele peak height was assessed using triplicate amplifications at each DNA input amount via calculating the average, standard deviation and Coefficient of Variation (CV) for each allele’s peak height. Coefficient of variation for the allele peak heights was then averaged per DNA input amount. Average CVs of ?0.350 were indicative of the desired level of reproducibility.
Inter-locus peak balance: Overall locus-to-locus balance, regardless of locus size, was assessed by calculating the sum of each locus’ peak height divided by the profile’s total sum of peak heights to obtain each locus’ locus:profile peak height ratio (or locus peak height to total sum of peak heights, LPH:TPH). Then, the coefficient of variation for each profile’s LPH:TPH was calculated and averaged for each template amount. Although there are no known guidelines regarding acceptable levels of LPH:TPH CV values, CV averages of ?0.350 were targeted during the development of each fast PCR method based on inter-locus balance obtained using standard Identifiler amplifications (3μl reaction volume).
Intra-locus peak balance: Intra-locus balance was assessed by calculating peak height ratios at heterozygous loci; these were averaged at each of the DNA input amounts, and additionally, average instances of PHR <50% per profile were calculated. It should be noted that these fast PCR procedures were being developed for use with single source, reference samples, therefore PHR tolerances of 50% are acceptable, compared to the traditional 60% that is typically utilized for casework samples to help identify mixtures [17].
Other amplification artifacts: Stutter (n-4, n+4, n-8), pull-up, -A, Non-Specific Amplification (NSA) and baseline noise were also assessed as part of the DNA input range determination. Only the peaks that were actually called using GeneMapper® ID were included in these analyses; locus specific stutter filters as defined within the software were used, but no other filters were applied. For each stutter, pull-up or -A peak detected, its peak height was divided by the peak height of the true allele from which it originated in order to calculate the percentage of that peak (defined as “percent stutter”, “percent pull-up” or “percent -A”, respectively). These values were averaged for each template amount assessed. Additionally, average instances of stutter, pull-up, -A, NSA and baseline peaks per profile were also calculated. At times it was difficult to differentiate between low-level NSA and elevated baseline, given that all profiles assessed originated from only two individuals.
Stochastic threshold: The stochastic assessment utilized the lowest DNA input amount from the sensitivity range down to the highest DNA input amount in which complete dropout consistently occurred. Data were utilized from the above DNA input range study, and ranges used for each protocol varied due the protocol’s performance with respect to sensitivity. All profiles were reanalyzed using the laboratory’s limit of detection (25rfu) and examined for extreme dropout probability [18] by counting the number of dropout instances where the surviving allele of a heterozygote locus is higher than the limit of detection. Then, using various potential stochastic thresholds, the number of dropout occurrences was calculated and graphed against each potential threshold to determine an acceptable stochastic threshold in which zero occurrences of dropout occurred.
Precision: Precision of allele sizing was assessed via 9947A amplification controls loaded in triplicate within a single injection on a detection plate (capillaries 6, 9 and 14), along with an Identifiler allelic ladder. This set was injected three times to account for intra- and inter-injection precision (n=9 for each of the 26 alleles in 9947A). Assessment criteria was based upon that described by Applied Biosystems - standard deviation of allele sizing must be
Stutter assessment: In addition to the limited stutter assessment that was performed on called stutter peaks from the sensitivity range data, a more in depth assessment of stutter was performed on the optimal DNA input range, the 25 optimization check samples (see below) and all 9947A positive amplification controls. For this analysis, stutter filters were not applied during GeneMapper® ID analysis, thereby allowing the detection of all peaks (including stutter) that occurred using a 25rfu threshold. Average percent stutter was calculated for n-4, n+4 and n-8 stutter for each locus and amplification method. Any stutter peak that overlapped with a true allele was excluded from this analysis, as were overlapping n-4 and n+4 peaks. If an n±4 peak from the first allele overlapped with an n-8 peak from the second allele, is was averaged with n±4 stutter only, not n-8. Stutter peaks coinciding with pull-up from another dye channel were also excluded.
Optimization check: Profiles obtained from 25 different samples (all difference donors) served as a small optimization check to assist with selecting the best fast PCR protocol. Since development of two of the fast PCR protocols (AmpliTaq Gold Fast and SpeedSTAR) experienced significant problems obtaining PHR consistently above 50% and there was some evidence that PHR <50% were associated with large amplicon-sized loci and/or large allele separation (i.e., most notably for AmpliTaq Gold Fast and the sample exhibiting the largest allele difference [24 bases], which happened to be at a large amplicon locus [D18S51]), individuals were selected with varying degrees of allele separation (differing by up to nine alleles, or 36 bases) at D18S51 and other large sized loci (e.g., D2S1338 and FGA). Thus, in addition to assessing PHR as discussed above for the optimal DNA input range determination, it was also assessed for signs of greater intra-locus imbalance due to the magnitude of allele separation and/or locus itself. Additionally, these profiles were assessed for allele concordance, profile completeness (percent full profiles and average percent alleles detected per sample), inter-locus balance (as discussed above) and first pass success rates. First pass success rate was defined as the percentage of passing profiles obtained during the first round of testing (i.e., without re-extraction, re-amplification, re-injection, etc.). Passing profiles must have all alleles detected at or above threshold, all PHR ?50%, no called stutter peaks >20%, no called pull-up peaks >20% and no -A; Table 7 for a complete list of guidelines.
| Criteria | Pass | Fail |
| % Alleles Detecteda | 100%, no signs of mixture | <100% |
| PHR at all heterozygous loci | >% 5 | <50% at any locus |
| Pull-up | < % 2 | >20% |
| Stutter (n-4, n+4) | < % 2 | >20% |
| Stutter (n-8) | Occurrences at>2 occurrences (<5%) or any occurrence >5% | |
| Trialleles and microvariantsb | None | Any occurrence |
| -A | None | Any occurrence |
| +A | None | Any occurrence |
| Elevated Baseline | Occurrences atOccurrences at >3 loci | |
| Non-specific Amplification | None | Any occurrence |
| Occurrences at >3 loci | >2 oversaturated peaks | |
| Migration | All allele calls correct | Any occurrence of poor migrationc |
| Injection Failure | None | Any occurrence |
| Loss of Resolutiond | None | Any occurrence |
| Spikes | £1 occurrencee | >1 occurrencef |
Table 7: First Pass Analysis Guidelines.
Occurrence” relates to a detected peak that is called using GeneMapper® ID.
aAt a 75rfu threshold
bMust be reprocessed to confirm; however, given that all samples that were tested either originated from a known source or were also processed using a current procedure for comparison purposes, these would only fail if non-concordant with the known profile or profile obtained from the current process
cThat results in OL or miscalled allele(s)
dPoor ILS or unresolved peaks
eOkay if in multiple dye channels but occur at the same base size
fAt different base sizes
RESULTS AND DISCUSSION
Optimization of low volume, fast PCR protocols
AmpliTaq gold fast PCR: For this product, low PHR (<50%) was a continued problem and ultimately was remedied by a relatively long PCR cycle (10sec denaturation, 45sec annealing, and 45sec extension) compared to some of the other products. Furthermore, poor PHR also prevented the use of a 2-step PCR cycle. Final extension was a little longer than some of the other protocols (13min) in order to prevent -A, which still formed on occasion using a 10min final extension. The total run time for PCR on the Veriti was 1hr 19min - a 51% reduction from that of standard Identifiler using a 3µl reaction. Based on the data that utilized the optimized protocol (n=18), full profiles were obtained from all samples (0.25-0.75ng) with an average peak height of 891rfu, an average PHR of 83.6%, 17% of samples (limited to 0.25ng) exhibiting one or two PHR <50%, an average LPH:TPH CV value of 0.268 and no -A or NSA.
KAPA2G™ fast multiplex PCR kit: This product performed well from the start with the 3µl Identifiler amplification. The main obstacle to overcome using KAPA2G was the presence of -A, which was remedied by a 10min final extension. The next goal was to reduce overall amplification time without compromising STR profile quality. Ultimately, a 2-step PCR cycle with combined annealing/extension at 61?C was selected, which had a total run time of 43min - a 73% reduction from standard Identifiler using a 3µl reaction. Based on the data that utilized the optimized protocol (n=24), full profiles were obtained from all samples with an average peak height of 821rfu, an average PHR of 86.8%, 4% of samples exhibiting a single PHR <50%, an average LPH:TPH CV value of 0.294 and no -A or NSA.
Speed STAR™ HS DNA polymerase: Like AmpliTaq Gold Fast, Speed STAR also suffered from low PHR (<50%) and ultimately, profiles could not always be obtained with all PHR >50%. A longer denaturation time was attempted, as suggested by Giese et al., [2] and Walsh et al., [20], as were a 3-step PCR cycle (which helped to remedy low PHR with AmpliTaq Gold) and longer annealing/extension times (which was helpful with KAPA2G), but none of these, or other potential solutions, resolved the frequently low PHR. Similar to AmpliTaq Gold Fast as well, -A was not always prevented using a 10min final extension, so 13min was utilized. Comparatively, this optimized protocol was among the fastest - at 49min (a70% reduction compared to standard 3µl Identifiler) - and easily yielded full profiles well above analysis threshold (75rfu). Based on the data that utilized the optimized protocol (n=18), full profiles were obtained from all samples (0.25-0.75ng) with an average peak height of 1195rfu, an average PHR of 83.6%, 6% of samples (limited to 0.25ng) exhibiting a single PHR <50%, an average LPH:TPH CV value of 0.325 and no -A or NSA.
Type-it® microsatellite PCR kit: Unlike AmpliTaq Gold and Speed STAR, the Type-it kit exhibited better PHR using a 2-step PCR cycle compared to a 3-step, but it was very difficult to reduce amplification time because most attempts to reduce denaturation or combined anneal/extension either resulted in abundant allelic dropout and reduced peak height, or an increase in inter-locus imbalance. Thus, the optimized protocol was longer than hoped, at 1hr 14min - a 54% reduction in amplification time compared to standard 3µl Identifiler reactions. Based on the data that utilized the optimized protocol (n=18), full profiles were obtained from all samples (0.25-0.75ng) with an average peak height of 1209rfu, an average PHR of 84.0%, 6% of samples (limited to 0.25ng) exhibiting a single PHR <50%, an average LPH:TPH CV value of 0.329 and no -A or NSA.
Comparison of four low volume, fast PCR Protocols to low volume, standard PCR
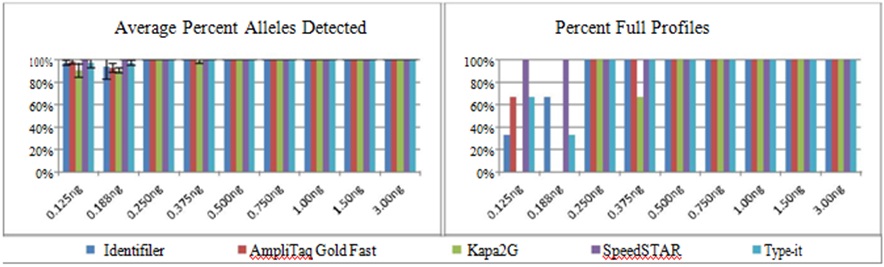
Determination of the optimal DNA input ranges: The optimal DNA input range was determined for each of the five amplication protocols (Figure 1) based on the analyses below. The largest optimal range (0.250-1.50ng) was obtained from standard Identifiler reactions and fast PCR using KAPA2G. Though the fast PCR protocols using SpeedSTAR and Type-it were superior to the other methods with regard to sensitivity (full profiles were obtained from 0.125-3.00ng), SpeedSTAR was unable to generate profiles without PHR <50% using 0.250ng DNA.
 Figure 1: Optimal DNA Input Ranges for Standard Identifiler and Four Fast PCR Protocols.
Figure 1: Optimal DNA Input Ranges for Standard Identifiler and Four Fast PCR Protocols.An optimal range (red) was determined for each amplification using the Identifiler primer set based upon the evaluated criteria (n=3 samples per DNA input). As a reminder, optimal ranges did not have to exhibit “perfection” for all criteria tested, but the most weight was placed on sensitivity and intra-locus balance. For example, standard Identifiler demonstrated less than ideal reproducibility and inter-locus balance at 0.250ng and 1.50ng, respectively, but this was not enough to warrant the exclusion of those inputs from its optimal range.
Sensitivity: For all five data sets, >99% of alleles were detected and 100% full profiles were obtained when ?0.250ng DNA was amplified, except for a single allele below threshold using KAPA2G with 0.375ng DNA (Figure 2). This range is wider than that osberved by Vallone et al. (0.4-1.0ng DNA) using 10μl fast PCR reactions consisting of a combination of PyroStart (Fermentas, Glen Burnie, MD) and SpeedSTAR polymerases [8], but lower than that observed by Foster and Laurin (0.125-2.0ng DNA) using 15μl fast PCR reactions with the SpeedSTAR polymerase [1].
Reproducibility: Allele peak height and balance is summarized in figure 3. As expected, average allele peak height increased as DNA input increased and tended to be higher using SpeedSTAR and Type-it compared to the other three methods. Reproducibility of peak height was measured via average CV per DNA input, which was ?0.350 when ?0.250ng DNA was amplified using each of the four fast PCR protocols, but was often >0.350 using standard Identifiler amplification, indicating less reproducibility using standard amplification compared to fast.
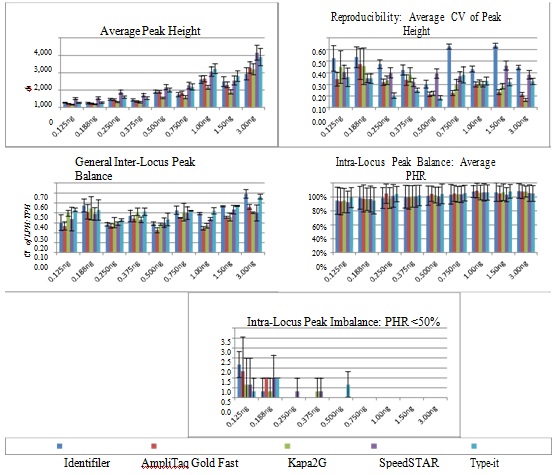 Figure 3: Peak Height Summary for Standard Identifiler and Four Fast PCR Protocols.
Figure 3: Peak Height Summary for Standard Identifiler and Four Fast PCR Protocols.Inter-locus Peak Balance: None of the five methods tested were able to consistently result in the desired level of general inter-locus balance (CV of LPH:TPH ?0.350), but balance for each of the fast PCR methods was about as good as or better than standard PCR when ?0.250ng DNA was amplified (Figure 3). It should be noted that CV increased to undesirable levels for all methods when 3.00ng was amplified.
Average peak height, reproducibility of peak height per allele (CV of PH), inter-locus peak balance (CV of LPH:TPH) and intra-locus peak balance/imbalance (average PHR and average number of PHR <50% per profile) are displayed for each DNA input (n=3 samples per DNA input).
Intra-locus Peak Balance: Intra-locus balance was measured via average heterozygote peak height ratios, which expectedly increased as DNAinput increased (Figure 3). Instances of PHR <50% tended to occur when ?0.188ng DNA was amplified for any of the five PCR methods and rarely occurred with higher DNA inputs.
Other Amplification Artifacts: Of the various artifacts that were observed above threshold, stutter (n-4) was the most prevalent (Figure 4) for all methods. Average percent stutter ranged from 9.7-17% and did not differ much based on DNA input. Instances of stutter increased as DNA input increased, tending to be more prevalent using AmpliTaq Gold Fast and SpeedSTAR, but never occurring using standard PCR with 20%) occurred occassionally, but were limited to profiles obtained with AmpliTaq Gold Fast or SpeedSTAR. Other forms of stutter (n+4 and n-8) did occur, but these peaks were not above analysis threshold, except for a single n-8 stutter peak obtained using SpeedSTAR and 3.00ng DNA (data not shown).
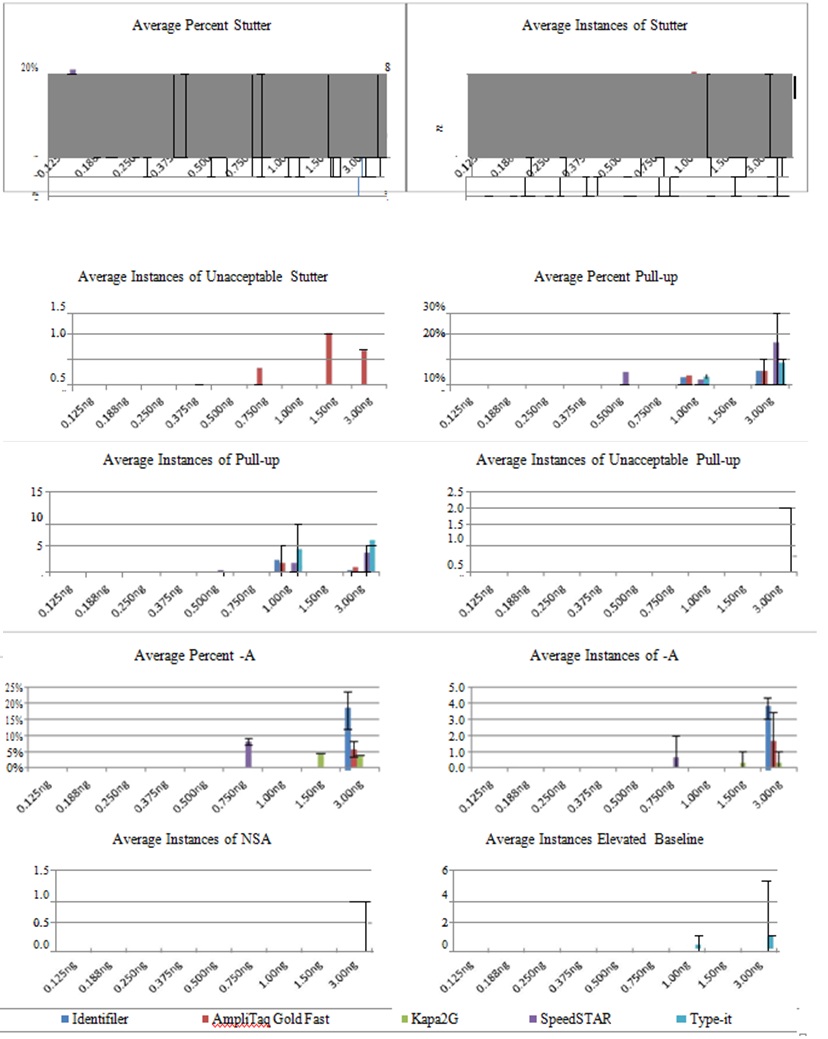 Figure 4: Artifacts for Standard Identifiler and Four Fast PCR Protocols.
Figure 4: Artifacts for Standard Identifiler and Four Fast PCR Protocols.Average percent stutter (n-4), pull-up and -A are displayed, as well as average number of detected stutter (n-4), pull-up, -A, low-level NSA and elevated baseline per profile. Average number of unacceptable stutter (n-4) and pull-up (i.e., those >20% of the true allele) per profile also displayed (n=3 samples per DNA input).
Pull-up peaks were the next most abundant artifact that were detected above threshold, but were nearly always limited to amplification of ?1.00ng DNA. Pull-up was more frequent for SpeedSTAR and Type-it, which alsohad higher percent pull-up than the other methods. These two methods were also the only ones to generate profiles with unacceptably high pull-up (percent pull-up >20%), but this was limited to amplification of 3.00ng DNA. This was likely associated with the higher average peak heights obtained using SpeedSTAR and Type-it. Other artifacts included -A, possible low-level non-specific amplification peaks and elevated baseline, but these were infrequent and tended to be limited to amplification of 3.00ng DNA.
Stochastic threshold: Stochastic thresholds were determined for each fast PCR method in order to identify a threshold at which heterozygous loci would not be mistaken for homozygous in the event of allelic drop out; these were compared to that of standard PCR (Figure 5). Fast PCR stochastic thresholds ranged from 75rfu (KAPA2G) to 165rfu (AmpliTaq Gold Fast), compared to 100rfu for standard PCR. It was not surprising that AmpliTaq Gold Fast most closely resemebled standard Identifiler, given that AmpliTaq Gold polymerase was used in that amplification.
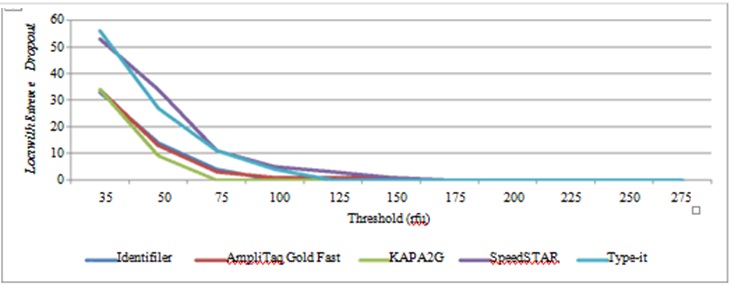 Figure 5: Stochastic Thresholds for Standard Identifiler and Four Fast PCR Protocols.
Figure 5: Stochastic Thresholds for Standard Identifiler and Four Fast PCR Protocols.Thresholds were established for each amplification method by determing the point at which heterzygous loci would not be mistaken for homozygous loci due to drop out of a single allele (n=447 to 522 loci for each amplification method).
Precision: Precision of allele sizing was assessed based on multiple injections of positive control 9947A amplified using each of the four fast PCR methods and compared to that of standard Identifiler. All assessments indicated acceptable levels of precision per manufacture recommendations (standard deviation <0.15; Figure 6).
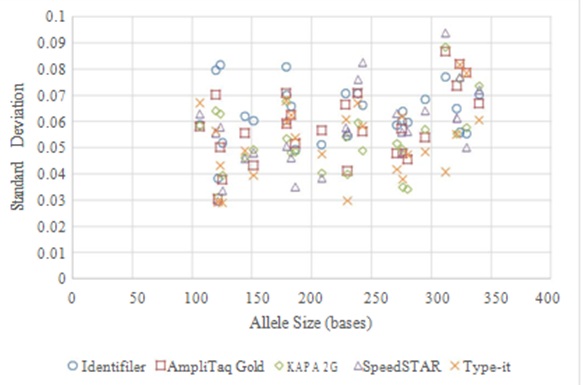 Figure 6: Precision of Allele Sizing for Standard Identifiler and Four Fast PCR Protocols.
Figure 6: Precision of Allele Sizing for Standard Identifiler and Four Fast PCR Protocols.Precision was assessed via standard deviation for each allele from 9947A positive control DNA (for each amplification method, n=9 for each of the 26 alleles). The upper limit of acceptable standard deviation is 0.15, and all protocols exhibit values well below that point.
Stutter assessment: For the detailed stutter analysis, n-4 stutter was the most frequent and accounted for 85% (SpeedSTAR) to 97% (KAPA2G) of all stutter peaks. Percent stutter (n-4) varied by amplification method (lowest from standard Identifiler and highest from AmpliTaq Gold Fast) and locus (tended to be lowest at TH01 and TPOX and highest at D21S11, D3S1358, D2S1338 and D18S51) (Figure 7). Use of a global 20% n-4 stutter allowance for all loci is common practice for reference samples [6] and would work well for any of the developed fast PCR methods. It should be noted, however, that the maximum observed percent stutter (n-4) was above 20% for all fast PCR methods, ranging from 20-26%. These were attributed to a single sample at locus D18S51. Comparatively, this sample exhibited an 18% stutter peak when amplified with standard Identifiler.
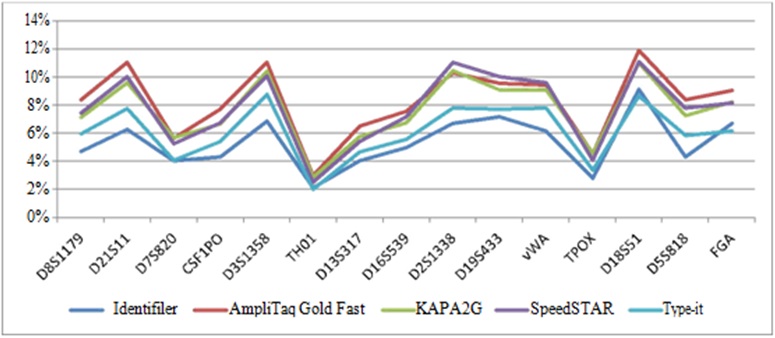
Both n+4 and n-8 stutter occurred significantly less, often not occurring at many loci (data not shown). Average n+4 percent stutter ranged from 3% (standard Identifiler, AmpliTaqGold Fast and SpeedSTAR) to 5% (KAPA2G and Type-it), whereas average n-8 percent stutter ranged from 1% (Type-it) to 2% (all other methods).
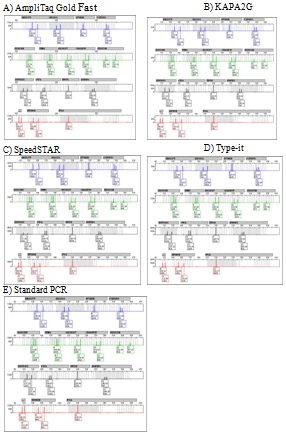
Optimization check: Twenty-five samples were processed using each of the four fast PCR methods, and the resulting profiles were compared to those obtained from standard Identifiler amplification. Full profiles were obtained from 96% of the samples using each of the fast PCR methods, compared to 92% using standard PCR, with an average of 98% alleles detected using each of the fast PCR methods and 96% of alleles using standard PCR; all profiles exhibited concordant allele calls and no unexplained alleles were present. Inter-locus peak balance was assessed via the average CV of LPH:TPH and was <0.350 for all methods except fast PCR using Type-it (average 0.380). Intra-locus peak balance was assessed via average PHR, occurrences of PHR <50% and signs of greater imbalance due to the magnitude of allele separation and/or locus. All methods exhibited average PHR between 2.7% and 84.7%, and all methods exhibited at least one profile with a single PHR <50%. Standard and fast PCR with KAPA2G exhibited 4% of profiles (i.e., one sample) with a single PHR <50%, AmpliTaq Gold Fast and Type-it each exhibited 8% of profiles with PHR <50%, while SpeedSTAR exhibited 32% of profiles with PHR <50% for one or more loci. When examined for increased intra-locus imbalance based on the magnitude of allele base-pair difference and/or locus, each of the five methods exhibited statistically significant differences with regard to one or both of these criteria (two-way ANOVA, α=0.05). However, when the data were further examined for correlation between PHR with either magnitude of allele difference or locus size, correlation was extremely weak (data not shown). Therefore, a decrease in PHR with regard to either an increase in the magnitude of allele repeat difference or an increase in locus size could not be substantiated for any of the amplification methods. Lastly, first pass success rates were assessed for each amplification method and ranged from 64% (SpeedSTAR) to 92% (KAPA2G), compared to 88% for standard Identifiler (as well as AmpliTaq Gold and Type-it); all failing profiles were due to either allelic dropout or PHR <50%. See Figure 8 for representative profiles from the five low volume protocols (four fast and one standard).
CONCLUSION
Low volume, fast PCR protocols for the Identifiler primer set were developed using four commercial products, but KAPA2G proved to be the most successful of the products that were evaluated. Furthermore, KAPA2G even had a higher first pass success rate than standard Identifiler using the same reaction volume, further demonstrating its suitability to produce high quality STR profiles from DNA reference samples.
But fast PCR is not all-perfect, especially when considering the different fast PCR products evaluated here. As has been demonstrated previously [1,5], percent stutter increases using each of the fast PCR protocols, as compared to that of standard PCR. However, use of a 20% global stutter filter or modification of the locus specific stutter thresholds in GeneMapper® ID should prevent excessive stutter peaks from being called by the software, which has already been demonstrated as an acceptable practice for reference DNA samples [6]. Vallone et al., (2008) reported more frequent occurrences of non-specific product formation for their 10μl Identifiler fast PCR protocol [8], which was only observed during the optimization phase of this study and not for any of the optimized protocols. Fast PCR does slightly reduce allele peak height, but using KAPA2G™ Fast Multiplex PCR Kit peak height is more reproducible than using standard Identifiler amplification (3µl reaction). Acceptable levels of intra- and inter-locus balance are obtainable using low volume, fast PCR, especially with KAPA2G. But for fast protocols presented here, slightly higher inter-locus imbalance was observed for all fast PCR compared to standard. Furthermore, the Speed STAR fast protocol frequently resulted in PHR <50%, which is highly undesirable.
This study also demonstrates that all of the fast polymerases evaluated have acceptable optimal amplifiable ranges for reference samples. KAPA2G has the widest optimal amplifiable range (0.250-1.50ng) compared to the other polymerases, especially to that of Speed STAR, which has the narrowest range (0.375-1.00ng). Comparatively, Foster and Laurin (2012) and Vallone et al., (2008) developed fast PCR protocols (25μl and 10μl, respectively) using the Identifiler primer set with Speed STAR and PyroStart/Speed STAR, respectively, that had optimal input ranges of 0.2-1ng and 0.4-1ng DNA [1,8]. It should be noted that the ranges between these studies are not directly comparable given the different quantification methods used, especially since that used in this study is not human specific.
Traditionally, most standard amplification protocols utilize a 4°C final hold, but this study demonstrates that a 25°C final hold is acceptable, as originally suggested by Foster and Laurin (2012) to prevent -A [1]. Using a higher temperature for the final hold does not have any adverse effects, though a reduction in -A formation may not be specifically seen (as was not in this study). However, it does result in a slightly shorter amplification (~1-2min shorter) and for some products, like AmpliTaq Gold Fast, results in an increase in peak height.
Fast PCR amplification was developed using the same thermal cycler (384-well Veriti) currently used in the laboratory selected for this project. Many laboratories are under budgetary constraints and cannot allocate funds for new and/or “fast” thermal cyclers, which are indeed costly. Therefore, it was a goal of this study to demonstrate to other laboratories that thermal cyclers that are commonly used in forensic DNA testing are suitable for fast PCR. Fast thermal cyclers can achieve shorter amplification times (as low as 19 min [2]) than the non-fast ones used here (43min to >1hr on the Veriti), but the need for such a short amplification may not be justified for a particular laboratory and, therefore, may not warrant such an expense.
Furthermore, this study set out to develop fast PCR protocols using a primer set that is commonly used in forensics in conjunction with a reasonably priced, commercially available fast polymerase. Again, this would demonstrate to other laboratories that there is not a need to have a specially concocted, home-brewed reaction or significantly increase reaction costs in order to achieve a shorter amplification process. KAPA2G™ Fast Multiplex PCR Kit is reasonably priced and resulted in a $0.06/sample cost increase for 3μl amplifications; for laboratories using full 25μl reactions, per sample costs will increase by $0.50, which could be a substantial amount depending on annual sample throughput. Despite having to purchase a fast PCR reagent in addition to the amplification kit, per sample amplification costs may actually remain the same or slightly decrease compared to the process utilized at Cellmark Forensics. Using their standard 3µl amplification process, AmpliTaq Gold® DNA polymerase is the limiting reagent for the Identifiler amplification kit, and implementing the KAPA2G fast PCR protocol will eliminate the need to purchase supplemental AmpliTaq Gold® DNA polymerase. This benefit could offset the cost of KAPA2G, but the extent of which remains to be determined.
In summary, there are no added costs due to supplies, labor or instrumentation for the low volume, fast PCR amplification process presented here with KAPA2G. Increases to reagent costs may be as high as $0.06/sample for 3μl reactions (or $0.50 for 25μl), but will likely be less given the elimination of the need to purchase supplemental AmpliTaq Gold® DNA polymerase that was necessary for standard amplification. Furthermore, KAPA2G fast PCR amplification results in a decrease of ~2hr of instrument usage. All-in-all, this study has demonstrated the development of robust, low volume fast PCR protocols for buccal samples using non-fast thermal cyclers with little to no increase in per sample costs.
FUTURE STUDIES
This was the first step towards developing low volume (3-6µl), fast PCR protocols for various primer sets commonly used in the forensic community using non-fast thermal cyclers. Now that the KAPA2G™ Fast Multiplex PCR Kit has demonstrated its suitability for low volume, fast PCR for DNA reference samples, future studies will include optimizing and validating a variety of primer sets commonly used in the forensic community that do not already include a fast polymerase - for example, the AmpF?STR® Identifiler® Plus PCR Amplification Kit and PowerPlex® 16 HS System primer sets. Coupling these protocols with non-fast thermal cyclers like the Veriti and GeneAmp® PCR System 9700, as well as testing them with a variety of reference sample types - including buccal swabs, buccal collectors, and blood cards - should also be pursued to make low volume, fast PCR more versatile for DNA reference samples.
ACKNOWLEDGEMENT
We would like to thank Takara Bio, Kapa Biosystems, Applied Biosystems and QIAGEN for donating reagents for this study.
CONFLICTS OF INTEREST
At the time this research was conducted, Catherine Connon and Aaron LeFebvre were employees of Cellmark Forensics, the sister lab to Bode Technology (now collectively known as Bode Cellmark Forensics). Reagents were donated for evaluation by several manufacturers (Takara Bio, Kapa Biosystems, Applied Biosystems and QIAGEN), but did not result in bias.
ROLE OF FUNDING SOURCE
This project was funded by Cellmark Forensics, a LabCorp Specialty Testing Group. Additional reagents were donated by Takara Bio, Kapa Biosystems, Applied Biosystems and QIAGEN.
REFERENCES
- Foster A, Laurin N (2012) Development of a fast PCR protocol enabling rapid generation of AmpF?STR® Identifiler® profiles for genotyping of human DNA. Investig Genet 3: 6.
- Giese H, Lam R, Selden R, Tan E (2009) Fast multiplexed polymerase chain reaction for conventional and microfluidic short tandem repeat analysis. J Forensic Sci 54: 1287-1296.
- Gray K, Crowle D, Scott P (2014) Direct amplification of casework bloodstains using the Promega PowerPlex(®) 21 PCR amplification system. Forensic Sci Int Genet 12: 86-92.
- Hedman J, Albinsson L, Ansell C, Tapper H, Hansson O, et al. (2008) A fast analysis system for forensic DNA reference samples. Forensic Sci Int Genet 2: 184-189.
- Laurin N, Frégeau C (2012) Optimization and validation of a fast amplification protocol for AmpFlSTR® Profiler Plus® for rapid forensic human identification. Forensic Sci Int Genet 6: 47-57.
- Myers BA, King JL, Budowle B (2012) Evaluation and comparative analysis of direct amplification of STRs using PowerPlex® 18D and Identifiler® Direct systems. Forensic Sci Int Genet 6: 640-645.
- Park SJ, Kim JY, Yang YG, Lee SH (2008) Direct STR amplification from whole blood and blood- or saliva-spotted FTA without DNA purification. J Forensic Sci 53: 335-341.
- Vallone PM, Hill CR, Butler JM (2008) Demonstration of rapid multiplex PCR amplification involving 16 genetic loci. Forensic Sci Int Genet 3: 42-45.
- Verheij S, Harteveld J, Sijen T (2012) A protocol for direct and rapid multiplex PCR amplification on forensically relevant samples. Forensic Sci Int Genet 6: 167-175.
- Wang DY, Chang CW, Hennessy LK (2009) Rapid STR analysis of single source DNA samples in 2 h. Forensic Sci Int Genet Suppl Ser 2: 115-116.
- Leclair B, Sgueglia JB, Wojtowicz PC, Juston AC, Frégeau CJ, et al. (2003) STR DNA typing: increased sensitivity and efficient sample consumption using reduced PCR reaction volumes. J Forensic Sci 48: 1001-1013.
- Federal Bureau of Investigation (2011) Quality Assurance Standards for Forensic DNA Testing Laboratories, effective 9-1-2011. Federal Bureau of Investigation, Washington DC, USA.
- Federal Bureau of Investigation (2011) Quality Assurance Standards for DNA Databasing Laboratories. Federal Bureau of Investigation, Washington DC, USA.
- Gangano S, Elliot K, Anoruo K, Gass J, Buscaino J (2013) DNA investigative lead development from blood and saliva samples in less than two hours using the RapidHIT™ Human DNA Identification System. Forensic Sci Int Genet Suppl Ser 4: 43-44.
- LaRue BL, Moore A, King JL, Marshall PL, Budowle B (2014) An evaluation of the RapidHIT(®) system for reliably genotyping reference samples. Forensic Sci Int Genet 13: 104-111.
- Invitrogen™ Life Technologies (2005) Instruction Manual: ChargeSwitch® Forensic DNA Purification Kits, Version A. Invitrogen Corporation, Carlsbad, CA, USA.
- Federal Bureau of Investigation (2010) SWGDAM Interpretation Guidelines for Autosomal STR Typing by Forensic DNA Testing Laboratories. Federal Bureau of Investigation, Washington DC, USA.
- Gill P, Puch-Solis R, Curran J (2009) The low-template-DNA (stochastic) threshold--its determination relative to risk analysis for national DNA databases. Forensic Sci Int Genet 3: 104-111.
- Life Technologies (2014) POP-4® Polymer for 3130/3130xl Genetic Analyzers. Invitrogen Corporation, Carlsbad, CA, USA.
- Walsh PS, Erlich HA, Higuchi R (1992) Preferential PCR amplification of alleles: mechanisms and solutions. PCR Methods Appl 1: 241-250.
Citation: Connon CC, LeFebvre AK, Benjamin RC (2016) Validation of Low Volume, Fast PCR Amplification of STR Loci for Reference DNA Samples. J Forensic Leg Investig Sci 2: 009.
Copyright: © 2016 Catherine C Connon, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.