Journal of Psychiatry Depression & Anxiety Category: Clinical
Type: Research Article
Pharmacogenomics of Antidepressants
*Corresponding Author(s):
Ramón CacabelosEuroespes Biomedical Research Center, Institute Of Medical Science And Genomic Medicine, Camilo José Cela University, 15165 Bergondo, A Coruña, Spain
Tel:+34 981780505, +34 981780511
Email:rcacabelos@euroespes.com
Received Date: Jul 14, 2015
Accepted Date: Aug 25, 2015
Published Date: Sep 15, 2015
Abstract
Depression is a major problem of mental health with a high prevalence in the community, and antidepressants are among the most prescribed drugs in developed countries. Pharmacogenomics accounts for over 60% variability in the pharmacodynamics and pharmacokinetics of antidepressants (selective serotonin reuptake inhibitors, serotonin-norepinephrine reuptake inhibitors, tricyclic and tetracyclic compounds, monoamine oxidase inhibitors, and noradrenergic and serotonergic modulators). The genes involved in the pharmacogenomic response to antidepressant drugs fall into five major categories: (i) genes associated with the pathogenesis of depression (disease-specific genes, pathogenic genes); (ii) genes associated with the mechanism of action of drugs (mechanistic genes); (iii) genes associated with drug metabolism (metabolic genes); (iv) genes associated with drug transporters; and (v) pleiotropic genes involved in multifaceted cascades and metabolic reactions. About 24% of antidepressants are major substrates of CYP1A2 enzymes, 5% of CYP2B6, 38% of CYP2C19, 85% of CYP2D6, and 38% of CYP3A4. Among Caucasians, approximately one-quarter of the population is deficient in the enzymatic activity of the CYP2D6-CYP2C19-CYP2C9 cluster responsible for the metabolism of over 60% of current drugs. The implementation of pharmacogenomic procedures in the clinical setting would help to optimize the use of antidepressants in psychiatric patients.
Keywords
APOE; CYPs; Antidepressants; Depression; Pharmacogenomics
INTRODUCTION
Depression is a major problem of mental health in the community with a prevalence of 5-10% for females and 2-5% for males, and a lifetime risk of 10-25% in women and 5-12% in men. Antidepressants are among the most prescribed drugs in the USA and the EU. The most important classes of antidepressants are the Selective Serotonin Reuptake Inhibitors (SSRIs), Serotonin-Norepinephrine Reuptake Inhibitors (SNRIs), Tricyclic and Tetracyclic Antidepressants (TCAs), Monoamine Oxidase Inhibitors (MAOIs), and Noradrenergic and Serotonergic Modulators [1].
Since the pioneering works of Bönicke and Reif, Carson and coworkers, Kalow and Staron, and Motulsky in the 1950s, and the introduction of the concept of “pharmacogenetics” by Vogel in 1959, Sjöqvist and coworkers soon made clear in 1967-1973 that the metabolism of tricyclic antidepressants was genetically controlled [2-5]. Over 500 papers corroborated this assumption during the past half century. However, pharmacogenetics is still in its infancy and its concept has evolved into a broader spectrum after the completion of the human genome project [6-8]. At the present time, pharmacogenomics relates to the application of genomic technologies, such as genotyping, gene sequencing, gene expression, genetic epidemiology, transcriptomics, proteomics, metabolomics and bioinformatics, to drugs in clinical development and on the market, applying the large-scale systematic approaches of genomics to speed up the discovery of drug response markers, whether they act at the level of drug target, drug metabolism, or disease pathways. For the past decade, several books have been published illustrating the progress of pharmacogenomics [3,9-14], culminating in the first World Guide for Drug Use and Pharmacogenomics published in 2012 [1].
While antidepressants are widely used to treat major depressive disorder and anxiety disorders, only half of the patients will respond to antidepressant treatment and only a third of patients will experience a remission of symptoms [15]. Both pharmacodynamics and pharmacokinetic properties of antidepressants and other CNS drugs are highly dependent upon pharmacogenomic factors [16]. In comparison with other pharmacological categories, the basic pharmacogenetics of antidepressants is relatively well known in vivo and in vitro. Most antidepressants are metabolized via CYP enzymes. CYP variants may potentially influence the metabolism of major antidepressants: amitriptyline, amoxapine, citalopram, clomipramine, desipramine, doxepin, duloxetine, escitalopram, fluoxetine, fluvoxamine, imipramine, isocarboxazid, L-tryptophan, maprotiline, minaprine, mirtazapine, moclobemide, nefazodone, nortriptyline, paroxetine, phenelzine, protriptyline, reboxetine, sertraline, tranylcypromine, trazodone, trimipramine, venlafaxine [1]. However, the genes involved in the pharmacogenomic response to antidepressant drugs may fall into five major categories: (i) genes associated with the pathogenesis of depression (disease-specific genes, pathogenic genes); (ii) genes associated with the mechanism of action of drugs (mechanistic genes); (iii) genes associated with drug metabolism (metabolic genes); (iv) genes associated with drug transporters (transporter genes); and (v) pleiotropic genes involved in multifaceted cascades and metabolic reactions [4,16-19]. Recent studies indicate that the prescription of antidepressants by trial-and-error, neglecting the pharmacogenetic profile of the patients, is subjected to an error rate over 60% with the consequent problems in efficacy and safety [20,21].
The therapeutic lessons obtained from pharmacogenetics in the past, as pointed out by Meyer in 2004 [2], can be the following: (i) all drug effects vary from person to person and all drug effects are influenced by genes; (ii) most drug responses are multifactorial; (iii) genetic polymorphisms of single genes, including mutations in coding sequences, gene duplications, gene deletions and regulatory mutations affect numerous drug-metabolizing enzymes, including several Cytochrome-P450 enzymes (CYPs), N-Acetyltransferases (NAT), Thiopurine-Methyltransferase (TPMT), and UDP-Glucuronosyltransferases (UDP-GT); individuals that possess these polymorphisms are at risk of experiencing documented adverse reactions or inefficacy of drugs at usual doses; (iv) genetic polymorphisms of drug targets and drug transporters are increasingly recognized (receptors, ion channels, growth factors) as causing variation in drug responses; (v) several targets respond to treatment only in subgroups of patients who carry sensitizing mutations of these targets; (vi) the frequency of variation of drug effects, whether multifactorial or genetic, varies considerably in ethnically defined populations; and (vii) application of response-predictive genetic profiles on clinical outcomes has, so far, been done mostly in academic centers and has not yet reached clinical practice [2].
To gain expertise in the pharmacogenomics of a particular disease it is necessary to understand the principles of 5 basic steps: (a) the genetics of the disorder to be studied in all its modalities (Mendelian genetics, susceptibility genetics, mitochondrial genetics, epigenetic phenomena, genome-environment interactions); (b) structural and functional genomics; (c) proteomics; (d) metabolomics; and (e) pharmacogenomics. The development of new compounds or retesting of old drugs by using pharmacogenetic strategies encompasses the following steps in a multidisciplinary fashion: (a) genetic screening (genotyping) of single genes to identify major gene targets; (b) analysis of genetic variation to differentiate populations; (c) structural and functional genomic analyses including genetic clusters and haplotypes; (d) analysis of genotype-phenotype correlations to characterize major phenotypes as therapeutic targets associated with a particular gene or a cluster of genes involved in a metabolic pathway; and (e) implementation of basic and clinical pharmacogenomics procedures for drug development [16,22-24].
Since the pioneering works of Bönicke and Reif, Carson and coworkers, Kalow and Staron, and Motulsky in the 1950s, and the introduction of the concept of “pharmacogenetics” by Vogel in 1959, Sjöqvist and coworkers soon made clear in 1967-1973 that the metabolism of tricyclic antidepressants was genetically controlled [2-5]. Over 500 papers corroborated this assumption during the past half century. However, pharmacogenetics is still in its infancy and its concept has evolved into a broader spectrum after the completion of the human genome project [6-8]. At the present time, pharmacogenomics relates to the application of genomic technologies, such as genotyping, gene sequencing, gene expression, genetic epidemiology, transcriptomics, proteomics, metabolomics and bioinformatics, to drugs in clinical development and on the market, applying the large-scale systematic approaches of genomics to speed up the discovery of drug response markers, whether they act at the level of drug target, drug metabolism, or disease pathways. For the past decade, several books have been published illustrating the progress of pharmacogenomics [3,9-14], culminating in the first World Guide for Drug Use and Pharmacogenomics published in 2012 [1].
While antidepressants are widely used to treat major depressive disorder and anxiety disorders, only half of the patients will respond to antidepressant treatment and only a third of patients will experience a remission of symptoms [15]. Both pharmacodynamics and pharmacokinetic properties of antidepressants and other CNS drugs are highly dependent upon pharmacogenomic factors [16]. In comparison with other pharmacological categories, the basic pharmacogenetics of antidepressants is relatively well known in vivo and in vitro. Most antidepressants are metabolized via CYP enzymes. CYP variants may potentially influence the metabolism of major antidepressants: amitriptyline, amoxapine, citalopram, clomipramine, desipramine, doxepin, duloxetine, escitalopram, fluoxetine, fluvoxamine, imipramine, isocarboxazid, L-tryptophan, maprotiline, minaprine, mirtazapine, moclobemide, nefazodone, nortriptyline, paroxetine, phenelzine, protriptyline, reboxetine, sertraline, tranylcypromine, trazodone, trimipramine, venlafaxine [1]. However, the genes involved in the pharmacogenomic response to antidepressant drugs may fall into five major categories: (i) genes associated with the pathogenesis of depression (disease-specific genes, pathogenic genes); (ii) genes associated with the mechanism of action of drugs (mechanistic genes); (iii) genes associated with drug metabolism (metabolic genes); (iv) genes associated with drug transporters (transporter genes); and (v) pleiotropic genes involved in multifaceted cascades and metabolic reactions [4,16-19]. Recent studies indicate that the prescription of antidepressants by trial-and-error, neglecting the pharmacogenetic profile of the patients, is subjected to an error rate over 60% with the consequent problems in efficacy and safety [20,21].
The therapeutic lessons obtained from pharmacogenetics in the past, as pointed out by Meyer in 2004 [2], can be the following: (i) all drug effects vary from person to person and all drug effects are influenced by genes; (ii) most drug responses are multifactorial; (iii) genetic polymorphisms of single genes, including mutations in coding sequences, gene duplications, gene deletions and regulatory mutations affect numerous drug-metabolizing enzymes, including several Cytochrome-P450 enzymes (CYPs), N-Acetyltransferases (NAT), Thiopurine-Methyltransferase (TPMT), and UDP-Glucuronosyltransferases (UDP-GT); individuals that possess these polymorphisms are at risk of experiencing documented adverse reactions or inefficacy of drugs at usual doses; (iv) genetic polymorphisms of drug targets and drug transporters are increasingly recognized (receptors, ion channels, growth factors) as causing variation in drug responses; (v) several targets respond to treatment only in subgroups of patients who carry sensitizing mutations of these targets; (vi) the frequency of variation of drug effects, whether multifactorial or genetic, varies considerably in ethnically defined populations; and (vii) application of response-predictive genetic profiles on clinical outcomes has, so far, been done mostly in academic centers and has not yet reached clinical practice [2].
To gain expertise in the pharmacogenomics of a particular disease it is necessary to understand the principles of 5 basic steps: (a) the genetics of the disorder to be studied in all its modalities (Mendelian genetics, susceptibility genetics, mitochondrial genetics, epigenetic phenomena, genome-environment interactions); (b) structural and functional genomics; (c) proteomics; (d) metabolomics; and (e) pharmacogenomics. The development of new compounds or retesting of old drugs by using pharmacogenetic strategies encompasses the following steps in a multidisciplinary fashion: (a) genetic screening (genotyping) of single genes to identify major gene targets; (b) analysis of genetic variation to differentiate populations; (c) structural and functional genomic analyses including genetic clusters and haplotypes; (d) analysis of genotype-phenotype correlations to characterize major phenotypes as therapeutic targets associated with a particular gene or a cluster of genes involved in a metabolic pathway; and (e) implementation of basic and clinical pharmacogenomics procedures for drug development [16,22-24].
PATHOGENIC GENES
The molecular mechanisms underlying Major Depressive Disorder (MDD) are largely unknown. Heritability of major depressive disorder is estimated to be at 0.36 to 0.70 [25], with the relative risk of the disorder 4-8 times greater in relatives of probands [26]. Over 1,000 different genes distributed across the human genome have been screened for major depression for the past decade, and less than 100 genes remain potentially associated with depression in different populations (Table 1). Classic genes conventionally associated with MDD (unipolar depression) include FKBP5 (FK506-Binding Protein 5) (6p21.31) (Major depressive disorder and accelerated response to antidepressant drug treatment) [27], TPH2 (Tryptophan Hydroxylase 2) (12q21.1) (susceptibility to unipolar depression and susceptibility to ADHD-2) [28], MDD1 (Major Depression Disorder 1) (12q22-q23.2) [29], HTR2A (5-Hydroxytryptamine Receptor 2A) (13q14.2) (response to citalopram therapy in major depressive disorder; susceptibility to alcohol dependence, anorexia nervosa, obsessive-compulsive disorder, schizophrenia, and seasonal affective disorder) [30], and MDD2 (Major Depressive Disorder 2, unipolar depression 2) (15q25.3-q26.2) [31]. Cytogenetic analysis also revealed that patients with a balanced translocation t (1;11) (q43;q21) may be at risk of suffering MMD and other mental disorders [32]. Heterozygous mutations in the DCTN1 gene on chromosome 2p13 have been associated with Perry syndrome, an autosomal dominant neurodegenerative disorder classically characterized by adult-onset Parkinsonism and depression, frontotemporal dementia and progressive supranuclear palsy. DCTN1 gene variants can also cause distal motor neuronopathy type VIIB and confer increased susceptibility to amyotrophic lateral sclerosis [33-34].

Table 1: Selected genes potentially associated with mood disorders.
Many other genes may be involved in the pathogenesis of depression [35,36] (Table 1). New players, such as epigenetic changes, alterations in mtDNA related to oxidative stress, and modifications in telomere length may also contribute to alter neuronal mechanisms involved in depressive conditions [37]. SNPs of genes involved in DNA repair, such as genes encoding three glycosylases (hOGG1, MUTYH and NEIL1) (c.977C>G - hOGG1 (rs1052133), c.972G>C - MUTYH (rs3219489) and c.*589G>C - NEIL1 (rs4462560)), particularly in the base excision repair (BER) pathway, may also modulate the risk of recurrent Depression (rDD). The C/C genotype and allele C of the c.*589G>C decrease the risk of rDD occurrence, while the G/G genotype and allele G of the same SNP increase the risk. This polymorphism has a stronger association with early-onset depression (C increases the risk of late-onset rDD, and the combined genotype C/C-C/C of c.977C>G and c.*589G>C significantly reduces the risk of rDD [38].
Genome-wide association studies have suggested a role for a genetic variation in the presynaptic gene PCLO in MDD. As with many complex traits, the PCLO variant has a small contribution to the overall heritability and the association does not always replicate. The PCLO p.Ser4814Ala missense variant produces mild cellular phenotypes, which do not translate into behavioral phenotypes [39]. Milaneschi et al., [40] examined the polygenic features of MDD and two common clinical subtypes (typical and atypical). MDD subtypes had differential polygenic signatures: typical was strongly associated with schizophrenia, while atypical was additionally associated with Body Mass Index (BMI) and triglycerides. SNP-heritability was 32% for MDD, 38% and 43% for subtypes with, respectively, decreased (typical) and increased (atypical) appetite/weight.
MECHANISTIC GENES
Most genes associated with the mechanism of action of antidepressant drugs encode receptors, enzymes, and neurotransmitters (serotonin, noradrenaline, dopamine, histamine, acetylcholine) on which drugs act as ligands (agonists, antagonists), enzyme modulators (substrates, inhibitors, inducers) or neurotransmitter regulators (releasers, reuptake inhibitors) [4].
METABOLIC GENES
Most pharmacogenetic studies with antidepressants are related to genes involved in drug metabolism. Excellent studies on the metabolism of antidepressants by different authors are collected in the World Guide for Drug Use and Pharmacogenomics [1]. Drug metabolism includes phase I reactions (i.e., oxidation, reduction and hydrolysis) and phase II conjugation reactions (i.e., acetylation, glucuronidation, sulphation and methylation). The principal enzymes with polymorphic variants involved in phase I reactions are the Cytochrome P450 monooxygenases (CYP3A4/5/7, CYP2E1, CYP2D6, CYP2C19, CYP2C9, CYP2C8, CYP2B6, CYP2A6, CYP1B1, CYP1A1/2), and other enzymes such as epoxide hydrolase, esterases, NQO1 (NADPH-Quinone Oxidoreductase), DPD (Dihydropyrimidine Dehydrogenase), ADH (Alcohol Dehydrogenase), and ALDH (Aldehyde Dehydrogenase); and major enzymes involved in phase II reactions include UGTs (Uridine 5'-Triphosphate Glucuronosyl Transferases), TPMT (Thiopurine Methyltransferase), COMT (Catechol-O-Methyltransferase), HMT (Histamine Methyl-Transferase), STs (Sulfotransferases), GST-A (Glutathione S-Transferase A), GST-P, GST-T, GST-M, NAT1 (N-Acetyl Transferase 1), NAT2, and others. Among these enzymes, CYP2D6, CYP2C9, CYP2C19, and CYP3A4/5 are the most relevant in the pharmacogenetics of Central Nervous System (CNS) drugs in general, and antidepressants in particular [1,4,17,18]. Approximately, 18% of neuroleptics are major substrates of CYP1A2 enzymes, 40% of CYP2D6, and 23% of CYP3A4; 24% of antidepressants are major substrates of CYP1A2 enzymes, 5% of CYP2B6, 38% of CYP2C19, 85% of CYP2D6, and 38% of CYP3A4; 7% of benzodiazepines are major substrates of CYP2C19 enzymes, 20% of CYP2D6, and 95% of CYP3A4. Most CYP enzymes exhibit ontogenic-, age-, sex-, circadian-, and ethnic-related differences. The practical consequence of this genetic variation is that the same drug can be differentially metabolized according to the genetic profile/expression during each subject’s lifespan, and that knowing the pharmacogenomic profile of an individual, his/her pharmacodynamic response is potentially predictable to some extent.
CYP2D6: CYP2D6 is a 4.38 kb gene with 9 exons mapped on 22q13.2. Four RNA transcripts of 1190-1684 bp are expressed in the brain, liver, spleen and reproductive system, where 4 major proteins of 48-55 kDa (439-494 aa) are identified. This protein is a transport enzyme of the cytochrome P450 subfamily IID or multigenic cytochrome P450 superfamily of mixed-function monooxygenases which localizes to the endoplasmic reticulum and is known to metabolize as many as 25% of commonly-prescribed drugs and over 60% of current psychotropics. The gene is highly polymorphic in the population. There are 141 CYP2D6 allelic variants, of which -100C>T, -1023C>T, -1659G>A, -1707delT, -1846G>A, -2549delA, -2613-2615delAGA, -2850C>T, -2988G>A, and -3183G>A represent the 10 most important variants. Different alleles result in the extensive, intermediate, poor, and ultra-rapid metabolizer phenotypes, characterized by normal, intermediate, decreased, and multiplied ability to metabolize the enzyme’s substrates, respectively. P450 enzymes convert xenobiotics into electrophilic intermediates which are then conjugated by phase II enzymes to hydrophilic derivatives that can be excreted. According to the database of the World Guide for Drug Use and Pharmacogenomics [1], 982 drugs are CYP2D6-related: 371 drugs are substrates, over 300 drugs are inhibitors, and 18 drugs are CYP2D6 inducers.
Among healthy individuals, Extensive Metabolizers (EMs) account for 55.71% of the population, whereas Intermediate Metabolizers (IMs) are 34.7%, Poor Metabolizers (PMs) 2.28%, and Ultra-Rapid Metabolizers (UMs) 7.31%. Among patients with depression, 64.76% are EMs, 27.31% are IMs, 4.85% are PMs, and 3.08 are UMs (Figures 1 and 2). Remarkable interethnic differences exist in the frequency of the PM and UM phenotypes among different societies all over the world. On average, approximately 6.28% of the world population belongs to the PM category. Europeans (7.86%), Polynesians (7.27%), and Africans (6.73%) exhibit the highest rate of PMs, whereas Orientals (0.94%) show the lowest rate. The frequency of PMs among Middle Eastern populations, Asians, and Americans is in the range of 2-3%. CYP2D6 gene duplications are relatively infrequent among Northern Europeans, but in East Africa the frequency of alleles with duplication of CYP2D6 is as high as 29%. In Europe, there is a North-South gradient in the frequency of PMs (6-12% of PMs in Southern European countries, and 2-3% PMs in Northern latitudes) [1,4,17]. In a recent study, Bagheri et al., [41] compared the prevalence of the CYP2D6*10, *4, and *14 alleles in an Iranian population of different ethnicities with those of other populations. The CYP2D6*4 (G1846A) and *14 (G1758A) allelic frequencies were not detected in different ethnicities, demonstrating the absence of a significant contribution of these alleles in Iranian populations. However, the T/T, C/T, and C/C genotype frequencies of the CYP2D6*10 allele were significantly different in all Iranian ethnic groups. The frequency of the homozygous T/T variant of the CYP2D6*10 allele was significantly high in the Lure and low in the Kurd ethnicities. The frequency of the T/T variant of the CYP2D6*10 allele in central Iran was the highest, while the south of Iran had the lowest frequency. About 39.3% of subjects (24.3% homozygous T/T CYP2D6*10 as PMs and 15% heterozygous C/T CYP2D6*10 as IMs) with this genotype are candidates to experience Adverse Drug Reactions (ADRs) with common drugs in Iran.
CYP2D6: CYP2D6 is a 4.38 kb gene with 9 exons mapped on 22q13.2. Four RNA transcripts of 1190-1684 bp are expressed in the brain, liver, spleen and reproductive system, where 4 major proteins of 48-55 kDa (439-494 aa) are identified. This protein is a transport enzyme of the cytochrome P450 subfamily IID or multigenic cytochrome P450 superfamily of mixed-function monooxygenases which localizes to the endoplasmic reticulum and is known to metabolize as many as 25% of commonly-prescribed drugs and over 60% of current psychotropics. The gene is highly polymorphic in the population. There are 141 CYP2D6 allelic variants, of which -100C>T, -1023C>T, -1659G>A, -1707delT, -1846G>A, -2549delA, -2613-2615delAGA, -2850C>T, -2988G>A, and -3183G>A represent the 10 most important variants. Different alleles result in the extensive, intermediate, poor, and ultra-rapid metabolizer phenotypes, characterized by normal, intermediate, decreased, and multiplied ability to metabolize the enzyme’s substrates, respectively. P450 enzymes convert xenobiotics into electrophilic intermediates which are then conjugated by phase II enzymes to hydrophilic derivatives that can be excreted. According to the database of the World Guide for Drug Use and Pharmacogenomics [1], 982 drugs are CYP2D6-related: 371 drugs are substrates, over 300 drugs are inhibitors, and 18 drugs are CYP2D6 inducers.
Among healthy individuals, Extensive Metabolizers (EMs) account for 55.71% of the population, whereas Intermediate Metabolizers (IMs) are 34.7%, Poor Metabolizers (PMs) 2.28%, and Ultra-Rapid Metabolizers (UMs) 7.31%. Among patients with depression, 64.76% are EMs, 27.31% are IMs, 4.85% are PMs, and 3.08 are UMs (Figures 1 and 2). Remarkable interethnic differences exist in the frequency of the PM and UM phenotypes among different societies all over the world. On average, approximately 6.28% of the world population belongs to the PM category. Europeans (7.86%), Polynesians (7.27%), and Africans (6.73%) exhibit the highest rate of PMs, whereas Orientals (0.94%) show the lowest rate. The frequency of PMs among Middle Eastern populations, Asians, and Americans is in the range of 2-3%. CYP2D6 gene duplications are relatively infrequent among Northern Europeans, but in East Africa the frequency of alleles with duplication of CYP2D6 is as high as 29%. In Europe, there is a North-South gradient in the frequency of PMs (6-12% of PMs in Southern European countries, and 2-3% PMs in Northern latitudes) [1,4,17]. In a recent study, Bagheri et al., [41] compared the prevalence of the CYP2D6*10, *4, and *14 alleles in an Iranian population of different ethnicities with those of other populations. The CYP2D6*4 (G1846A) and *14 (G1758A) allelic frequencies were not detected in different ethnicities, demonstrating the absence of a significant contribution of these alleles in Iranian populations. However, the T/T, C/T, and C/C genotype frequencies of the CYP2D6*10 allele were significantly different in all Iranian ethnic groups. The frequency of the homozygous T/T variant of the CYP2D6*10 allele was significantly high in the Lure and low in the Kurd ethnicities. The frequency of the T/T variant of the CYP2D6*10 allele in central Iran was the highest, while the south of Iran had the lowest frequency. About 39.3% of subjects (24.3% homozygous T/T CYP2D6*10 as PMs and 15% heterozygous C/T CYP2D6*10 as IMs) with this genotype are candidates to experience Adverse Drug Reactions (ADRs) with common drugs in Iran.
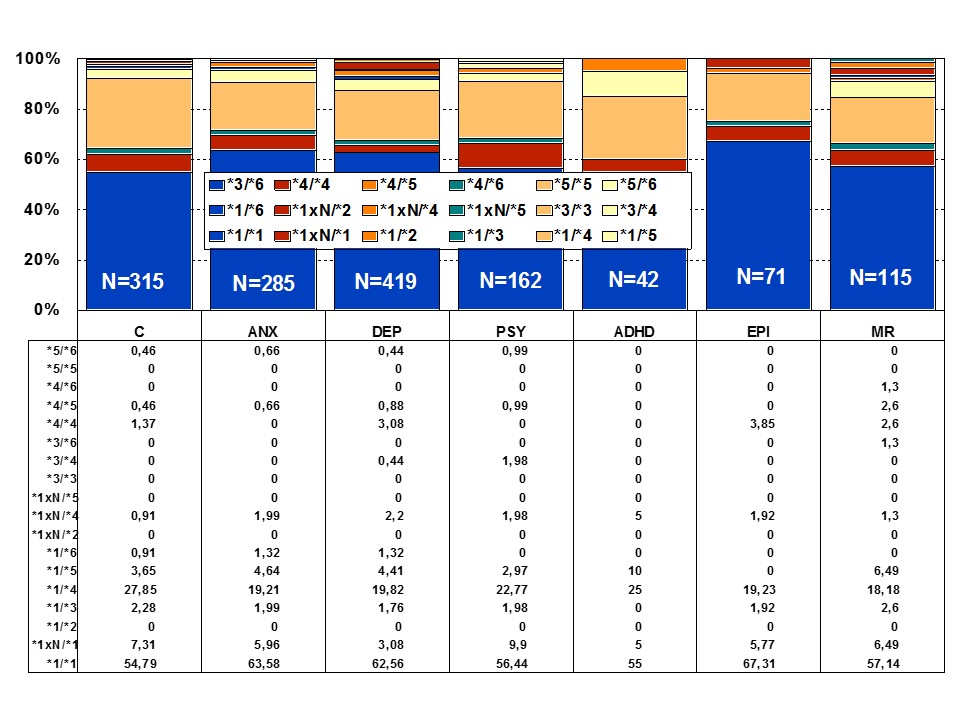 Figure 1: Distribution of CYP2D6 genotypes among patients with Anxiety (ANX), Depression (DEP), Psychosis (PSY), ADHD, Epilepsy (EPI), Mental Retardation (MR), and controls.
Figure 1: Distribution of CYP2D6 genotypes among patients with Anxiety (ANX), Depression (DEP), Psychosis (PSY), ADHD, Epilepsy (EPI), Mental Retardation (MR), and controls.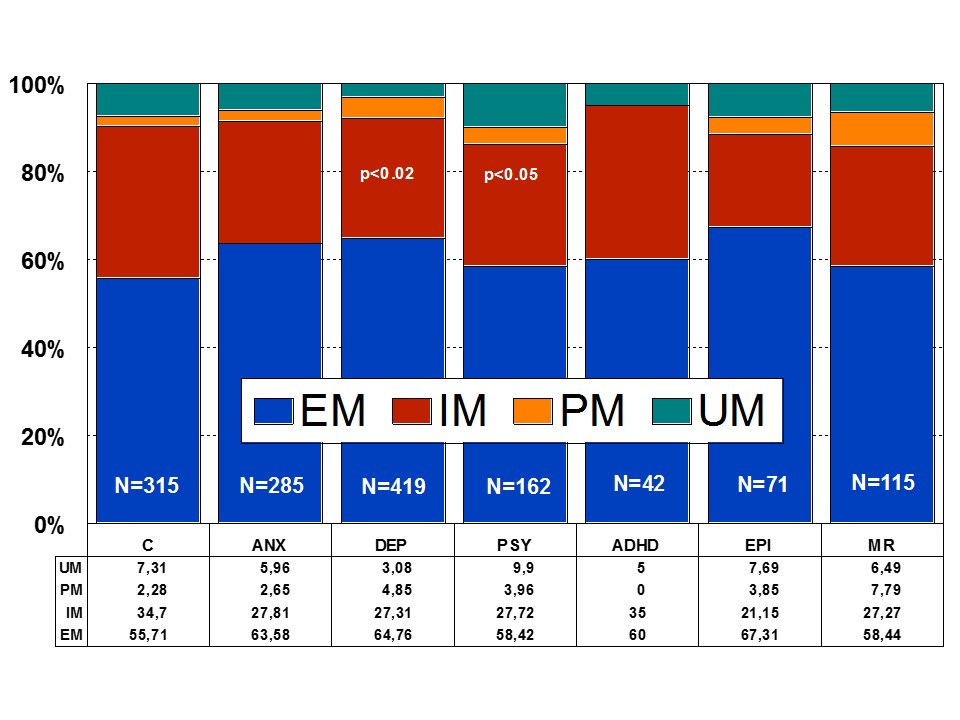
Figure 2: Distribution of CYP2D6 Extensive Metabolizers (EM), Intermediate Metabolizers (IM), Poor Metabolizers (PM), and Ultra-rapid Metabolizers (UM) among patients with Anxiety (ANX), Depression (DEP), Psychosis (PSY), ADHD, Epilepsy (EPI), Mental Retardation (MR), and controls.
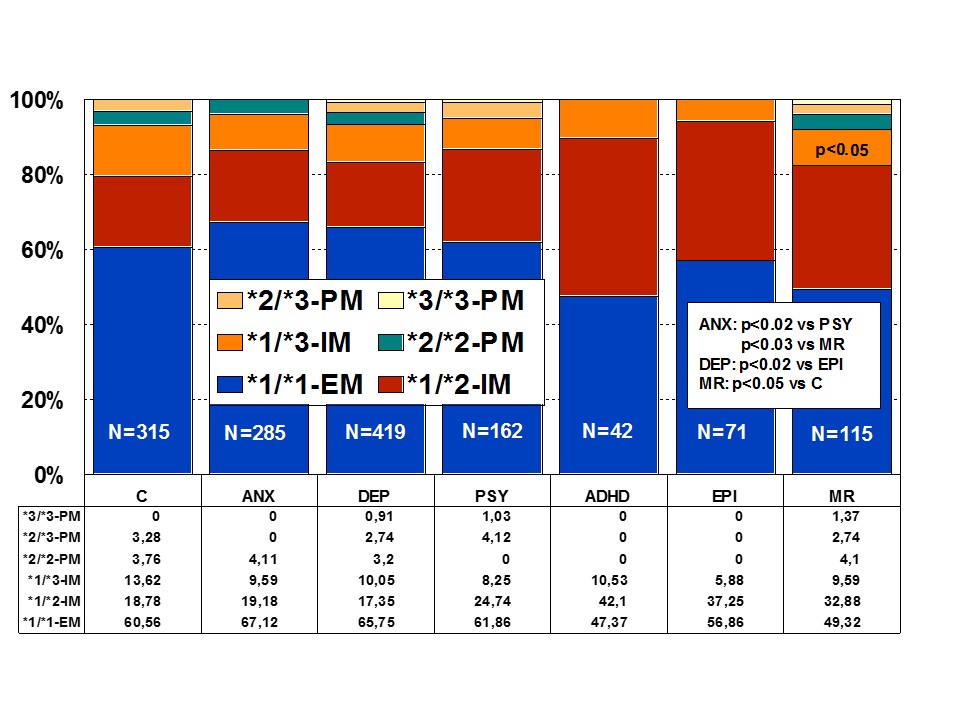
Figure 3: Distribution of CYP2C9 Extensive Metabolizers (EM), Intermediate Metabolizers (IM), and Poor Metabolizers (PM) among patients with Anxiety (ANX), Depression (DEP), Psychosis (PSY), ADHD, Epilepsy (EPI), Mental Retardation (MR), and controls.
CYP2C19: CYP2C19 is a gene (90.21 kb) with 9 exons mapped on 10q24.1q24.3. RNA transcripts of 1901 bp, 2395 bp, and 1417 bp are expressed in liver cells where a protein of 55.93 kDa (490 aa) is identified. Nearly 500 drugs are CYP2C19-related, 281 acting as substrates (151 are major substrates, 130 are minor substrates), 263 as inhibitors (72 weak, 127 moderate, and 64 strong inhibitors), and 23 as inducers of the CYP2C19 enzyme [1]. About 541 SNPs have been detected in the CYP2C19 gene. The frequencies of the 3 major CYP2C19 geno-phenotypes in the control population are CYP2C19-*1/*1-EMs 68.54%, CYP2C19-*1/*2-IMs 30.05%, and CYP2C19-*2/*2-PMs 1.41%. Minor variation has been reported in different CNS disorders [1,4,17] (Figure 4).
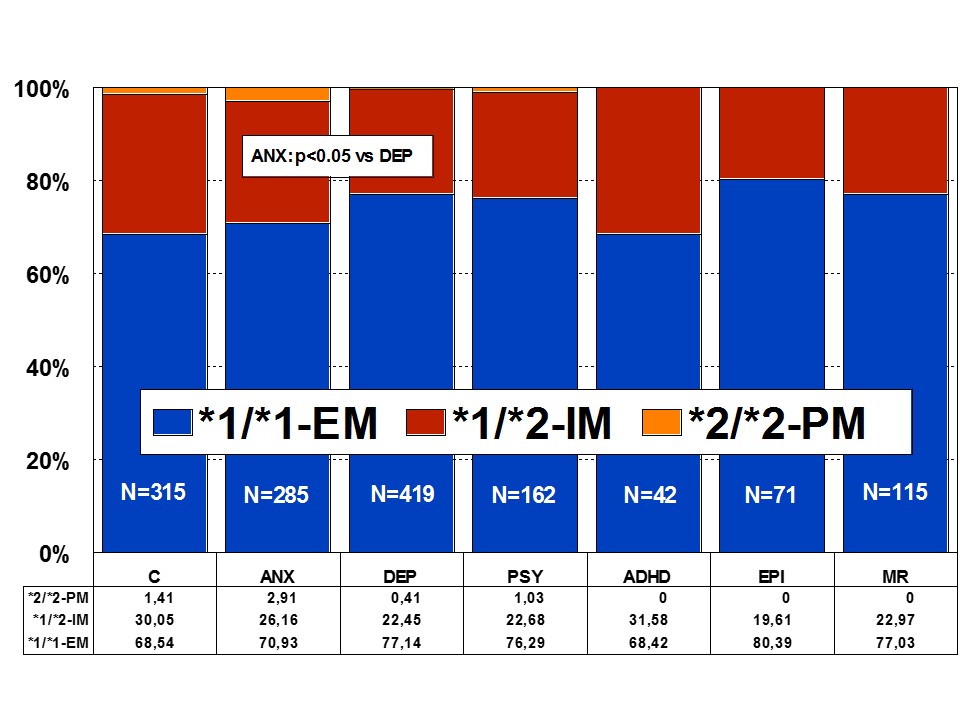
Figure 4: Distribution of CYP2C19 Extensive Metabolizers (EM), Intermediate Metabolizers (IM), and Poor Metabolizers (PM) among patients with Anxiety (ANX), Depression (DEP), Psychosis (PSY), ADHD, Epilepsy (EPI), Mental Retardation (MR), and controls.
CYP3A4/5: CYP3A4 is a gene (27.2 kb) with 13 exons mapped on 7q21.1. RNA transcripts of 2153 bp, 651 bp, 564 bp, 2318 bp and 2519 bp are expressed in intestine, liver, prostate and other tissues where 4 protein variants of 57.34 kDa (503 aa), 17.29 kDa (153 aa), 40.39 kDa (353 aa), and 47.99 kDa (420 aa) are identified. The human CYP3A locus contains the three CYP3A genes (CYP3A4, CYP3A5 and CYP3A7), three pseudogenes as well as a novel CYP3A gene termed CYP3A43. The gene encodes a putative protein with between 71.5% and 75.8% identity to the other CYP3A proteins. The predominant hepatic form is CYP3A4, but CYP3A5 contributes significantly to the total liver CYP3A activity. This enzyme metabolizes over 1900 drugs, 1033 acting as substrates (897 are major substrates, 136 are minor substrates), 696 as inhibitors (118 weak, 437 moderate, and 141 strong inhibitors), and 241 as inducers of the CYP3A4 enzyme [1]. About 347 SNPs have been identified in the CYP3A4 gene (CYP3A4*1A: Wild-type), 25 of which are of clinical relevance; in a Caucasian population, 82.75% are EMs (CYP3A5*3/*3), 15.88% are IMs (CYP3A5*1/*3), and 1.37% are UMs (CYP3A5*1/*1). Unlike other human P450s (CYP2D6, CYP2C19) there is no evidence of a ‘null’ allele for CYP3A4 [1].
CYP CLUSTERING
The construction of a genetic map integrating the most prevalent CYP2D6+CYP2C19+CYP2C9 polymorphic variants in a trigenic cluster yields 82 different haplotype-like profiles. The most frequent trigenic genotypes are *1*1-*1*1-*1*1 (25.70%), *1*1-*1*2-*1*2 (10.66%), *1*1-*1*2-*1*1 (10.45%), *1*4-*1*1-*1*1 (8.09%), *1*4-*1*2-*1*1 (4.91%), *1*4-*1*1-*1*2 (4.65%) and *1*1-*1*3-*1*3 (4.33%). These 82 trigenic genotypes represent 36 different pharmacogenetic phenotypes. According to these trigenic clusters, only 26.51% of the population show a pure 3EM phenotype, 15.29% are 2EM1IM, 2.04% are pure 3IM, 0% are pure 3PM, and 0% are 1UM2PM (the worst possible phenotype). This implies that only one-quarter of the population processes normally the drugs which are metabolized via CYP2D6, CYP2C9 and CYP2C19 (approximately 60% of the drugs of current use) [1,4,17].
TRANSPORTER GENES
ABC genes, especially ABCB1 (ATP-binding Cassette, subfamily B, member 1; P-glycoprotein-1, P-gp1; Multidrug Resistance 1, MDR1) (7q21.12), ABCC1 (9q31.1), ABCG2 (White1) (21q22.3), and other genes of this family encode proteins which are essential for drug metabolism and transport. The multidrug efflux transporters P-gp, Multidrug-Resistance associated Protein 4 (MRP4) and Breast Cancer Resistance Protein (BCRP), located on endothelial cells lining brain vasculature, play important roles in limiting movement of substances into and enhancing their efflux from the brain. Transporters also cooperate with Phase I/Phase II metabolism enzymes by eliminating drug metabolites. Their major features are their capacity to recognize drugs belonging to unrelated pharmacological classes, and their redundancy, by which a single molecule can act as a substrate for different transporters. This ensures an efficient neuroprotection against xenobiotic invasions. The pharmacological induction of ABC gene expression is a mechanism of drug interaction, which may affect substrates of the up-regulated transporter, and overexpression of MDR transporters confers resistance to anticancer agents and CNS drugs [42,43].
ABCB1 is probably the most important drug transporter in the brain. The ABCB1 gene maps on 7q21.12 spanning 209.39 kb (29 exons) with the structure of a P-glycoprotein and a Y-box sequence 5'-CTGATTGG-3' in its cis-regulatory elements. Several transcripts/variants are highly expressed in adrenal gland, blood-brain barrier (BBB), brain, kidney, liver, placenta, small intestine, and uterus, and low expression is present in many other tissues. These transcripts encode a protein (ABCB1-001: 141.48 kDa; 1280 aa. ABCB1-002: 5.89 kDa; 51 aa. ABCB1-003: 5.68 kDa; 48 aa. ABCB1-201: 2.52 kDa; 22 aa) of the ATP binding cassette superfamily, subfamily B (MDR/TAP) with two ATP binding and two Transmembrane (2TM) domains (2 x 6 segments), acting as a transport carrier and a lipid translocase of broad specificity. This is a large transmembrane protein which is an integral part of the BBB and functions as a drug-transport pump transporting a variety of drugs from the brain back into the blood. About 1630 ABCB1 variants have been identified [1]. Of interest, ABCB1 has approximately 116 polymorphic sites in Caucasians and 127 in African-Americans with a minor allele frequency greater than 5%. Some of the most commonly studied variants are 1236C>T, 2677G>A/T and 3435C>T and the most commonly studied haplotype involves the 1236, 2677 and 3435 (TTT) SNPs and 3 intronic SNPs (intron 9, intron 13, intron 14) named ABCB1*13. There are many other ABCB1 variants such as -129C>T (5'-UTR), 61A>G (Asn21Asp) and 1199G>A (Ser400Asn) that have been studied in vivo and in vitro. Variants of the ABCB1 gene have been associated with a diverse number of diseases and with a great variety of drugs, natural products and endogenous agents [1]. Over 1270 drugs have been reported to be associated with the ABCB1 transporter protein (P-gp), of which 490 are substrates, 618 are inhibitors, 182 are inducers, and 269 additional compounds which belong to different pharmacological categories of products with potential Abcb1 interaction [1].
Important for CNS pharmacogenomics are transporters encoded by genes of the Solute Carrier superfamily (SLC) and Solute Carrier Organic (SLCO) transporter family, responsible for the transport of multiple endogenous and exogenous compounds, including folate (SLC19A1), urea (SLC14A1, SLC14A2), monoamines (SLC29A4, SLC22A3), aminoacids (SLC1A5, SLC3A1, SLC7A3, SLC7A9, SLC38A1, SLC38A4, SLC38A5, SLC38A7, SLC43A2, SLC45A1), nucleotides (SLC29A2, SLC29A3), fatty acids (SLC27A1-6), neurotransmitters (SLC6A2 (noradrenaline transporter), SLC6A3 (dopamine transporter), SLC6A4 (serotonin transporter, SERT), SLC6A5, SLC6A6, SLC6A9, SLC6A11, SLC6A12, SLC6A14, SLC6A15, SLC6A16, SLC6A17, SLC6A18, SLC6A19), glutamate (SLC1A6, SLC1A7), and others [17]. Some Organic Anion Transporters (OAT), which belong to the Solute Carrier (SLC) 22A family, are also expressed at the BBB, and regulate the excretion of endogenous and exogenous organic anions and cations [44]. The transport of amino acids and di- and tripeptides is mediated by a number of different transporter families, and the bulk of oligopeptide transport is attributable to the activity of members of the SLC15A superfamily (Peptide Transporters 1 and 2 [SLC15A1 (PepT1) and SLC15A2 (PepT2)], and Peptide/Histidine Transporters 1 and 2 [SLC15A4 (PHT1) and SLC15A3 (PHT2)]). ABC and SLC transporters expressed at the BBB may cooperate to regulate the passage of different molecules into the brain [45]. Polymorphic variants in ABC and SLC genes may also be associated with pathogenic events in CNS disorders and drug-related safety and efficacy complications [4,16,17].
ABCB1 is probably the most important drug transporter in the brain. The ABCB1 gene maps on 7q21.12 spanning 209.39 kb (29 exons) with the structure of a P-glycoprotein and a Y-box sequence 5'-CTGATTGG-3' in its cis-regulatory elements. Several transcripts/variants are highly expressed in adrenal gland, blood-brain barrier (BBB), brain, kidney, liver, placenta, small intestine, and uterus, and low expression is present in many other tissues. These transcripts encode a protein (ABCB1-001: 141.48 kDa; 1280 aa. ABCB1-002: 5.89 kDa; 51 aa. ABCB1-003: 5.68 kDa; 48 aa. ABCB1-201: 2.52 kDa; 22 aa) of the ATP binding cassette superfamily, subfamily B (MDR/TAP) with two ATP binding and two Transmembrane (2TM) domains (2 x 6 segments), acting as a transport carrier and a lipid translocase of broad specificity. This is a large transmembrane protein which is an integral part of the BBB and functions as a drug-transport pump transporting a variety of drugs from the brain back into the blood. About 1630 ABCB1 variants have been identified [1]. Of interest, ABCB1 has approximately 116 polymorphic sites in Caucasians and 127 in African-Americans with a minor allele frequency greater than 5%. Some of the most commonly studied variants are 1236C>T, 2677G>A/T and 3435C>T and the most commonly studied haplotype involves the 1236, 2677 and 3435 (TTT) SNPs and 3 intronic SNPs (intron 9, intron 13, intron 14) named ABCB1*13. There are many other ABCB1 variants such as -129C>T (5'-UTR), 61A>G (Asn21Asp) and 1199G>A (Ser400Asn) that have been studied in vivo and in vitro. Variants of the ABCB1 gene have been associated with a diverse number of diseases and with a great variety of drugs, natural products and endogenous agents [1]. Over 1270 drugs have been reported to be associated with the ABCB1 transporter protein (P-gp), of which 490 are substrates, 618 are inhibitors, 182 are inducers, and 269 additional compounds which belong to different pharmacological categories of products with potential Abcb1 interaction [1].
Important for CNS pharmacogenomics are transporters encoded by genes of the Solute Carrier superfamily (SLC) and Solute Carrier Organic (SLCO) transporter family, responsible for the transport of multiple endogenous and exogenous compounds, including folate (SLC19A1), urea (SLC14A1, SLC14A2), monoamines (SLC29A4, SLC22A3), aminoacids (SLC1A5, SLC3A1, SLC7A3, SLC7A9, SLC38A1, SLC38A4, SLC38A5, SLC38A7, SLC43A2, SLC45A1), nucleotides (SLC29A2, SLC29A3), fatty acids (SLC27A1-6), neurotransmitters (SLC6A2 (noradrenaline transporter), SLC6A3 (dopamine transporter), SLC6A4 (serotonin transporter, SERT), SLC6A5, SLC6A6, SLC6A9, SLC6A11, SLC6A12, SLC6A14, SLC6A15, SLC6A16, SLC6A17, SLC6A18, SLC6A19), glutamate (SLC1A6, SLC1A7), and others [17]. Some Organic Anion Transporters (OAT), which belong to the Solute Carrier (SLC) 22A family, are also expressed at the BBB, and regulate the excretion of endogenous and exogenous organic anions and cations [44]. The transport of amino acids and di- and tripeptides is mediated by a number of different transporter families, and the bulk of oligopeptide transport is attributable to the activity of members of the SLC15A superfamily (Peptide Transporters 1 and 2 [SLC15A1 (PepT1) and SLC15A2 (PepT2)], and Peptide/Histidine Transporters 1 and 2 [SLC15A4 (PHT1) and SLC15A3 (PHT2)]). ABC and SLC transporters expressed at the BBB may cooperate to regulate the passage of different molecules into the brain [45]. Polymorphic variants in ABC and SLC genes may also be associated with pathogenic events in CNS disorders and drug-related safety and efficacy complications [4,16,17].
PLEIOTROPIC GENES
APOE is the prototypical paradigm of a pleiotropic gene with multifaceted activities in physiological and pathological conditions [46,47] (Figure 5). APOE variants are associated with dementia, cardiovascular disorders, and atherosclerosis [1] (Figure 5). There is an accumulation of APOE-4 carriers among patients with dementia, either degenerative or vascular [46] (Figure 5). ApoE is consistently associated with the amyloid plaque marker for Alzheimer’s Disease (AD). APOE-4 may influence AD pathology interacting with APP metabolism and Aβ accumulation, enhancing hyperphosphorylation of tau protein and NFT formation, reducing choline acetyltransferase activity, increasing oxidative processes, modifying inflammation-related neuroimmunotrophic activity and glial activation, altering lipid metabolism, lipid transport and membrane biosynthesis in sprouting and synaptic remodeling, and inducing neuronal apoptosis [22,46-49].
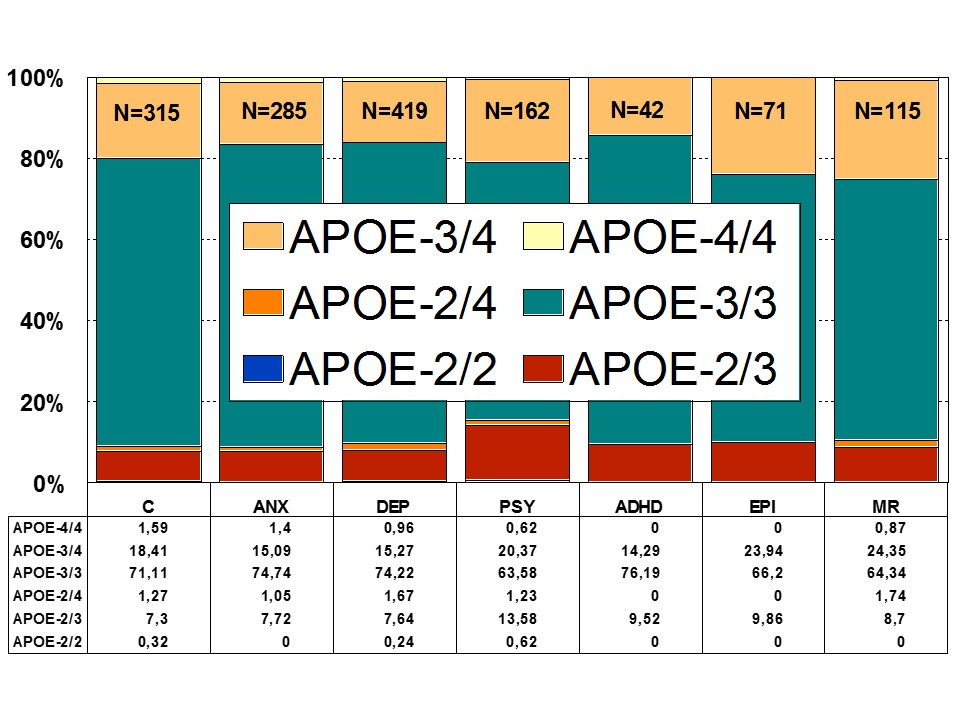 Figure 5: Distribution of APOE genotypes in patients with Anxiety (ANX), Depression (DEP), Psychosis (PSY), ADHD, Epilepsy (EPI), Mental Retardation (MR), and controls.
Figure 5: Distribution of APOE genotypes in patients with Anxiety (ANX), Depression (DEP), Psychosis (PSY), ADHD, Epilepsy (EPI), Mental Retardation (MR), and controls.An interactive effect of depressive symptoms and APOE e4 allele status on cognitive decline has been shown in old age. Carriers of the APOE-4 allele with more depressive symptoms have faster cognitive decline than those with either depression or the APOE-4 allele. Current depression is associated with poorer speed and memory. A negative effect of the APOE-4 allele on speed and memory is found in people older than 60 years of age [50] (Figure 5).
Influence of APOE and ACE on depression and anxiety in dementia
Behavioral disturbances and mood disorders are intrinsic components of dementia associated with memory disorders [51-55]. The appearance of anxiety, depression, psychotic symptoms, verbal and physical aggressiveness, agitation, wandering and sleep disorders complicate the clinical picture of dementia and add important problems to the therapeutics of AD and the daily management of patients as well. Under these conditions, psychotropic drugs (antidepressants, anxyolitics, hypnotics, and neuroleptics) are required, and most of these substances contribute to deteriorate cognition and psychomotor functions. Both APOE- and ACE-related polymorphic variants have been associated with mood disorders [56,57] and panic disorder [58]. Gender, age, dementia severity, APOE-4, and general medical health appear to influence the occurrence of individual neuropsychiatric symptoms in dementia, and medical comorbidity increases the risk of agitation, irritability, disinhibition, and aberrant motor behaviour [59]. A positive association between APOE-4 and neuropsychiatric symptoms [60] and depressive symptoms in AD has been reported [61], especially in women [62]. In other studies, no association of APOE-4 with behavioral dyscontrol (euphoria, disinhibition, aberrant motor behavior, and sleep and appetite disturbances), psychosis (delusions and hallucinations), mood (depression, anxiety, and apathy), and agitation (aggression and irritability) could be found [63]. Some authors did not find association of APOE-4 with major depression in AD [64,65] or in patients with major depression in a community of older adults [66], but an apparent protective effect of APOE-2 on depressive symptoms was detected [67]. Others, in contrast, found that APOE-4 was associated with an earlier age-of-onset, but not cognitive functioning, in late-life depression [68]. Apoe-/- mice without human ApoE or with APOE-4, but not APOE-3, show increased measures of anxiety [69]. Differences in anxiety-related behavior have been observed between APOE-deficient C57BL/6 and wild type C57BL/6 mice, suggesting that APOE variants may affect emotional state [70]. Histamine H3 autoreceptor antagonists increase anxiety measures in wilde-type mice, but not in ApoE-/- mice, and ApoE deficient mice show higher sensitivity to the anxiety-reducing effects of the H1 receptor antagonist mepyramine than wild-type mice, suggesting a role of H3-autoreceptor-mediated signaling in anxiety-like symptoms in this AD-related animal model [71].
In humans, APOE-4 carriers with deep white matter hyperintesities in Magnetic Resonance Imaging (MRI) show association with depressive symptoms and vascular depression [72]. Reduced caudate nucleus volumes and genetic determinants of homocysteine metabolism accumulate in patients with psychomotor slowing and cognitive deficits [73], and older depressed subjects have persisting cognitive impairments associated with hippocampal volume reduction [74,75]. Depressive symptoms are also associated with stroke and atherogenic lipid profile [76].
During the past two decades, antipsychotic, anti-anxiety and cognitive-enhancing effects have been attributed to ACE inhibitors [77,78]. It has been reported that some ACE inhibitors (captopril, SQ29,852) display similar effects to benzodiazepines in dealing with anxiety-related behaviors in animals [79], and another ACE inhibitor (ceronapril) might shared with neuroleptic drugs an ability to enhance latent inhibition in learning tasks [77]. One SNP (rs4291) located in the promoter region of the ACE gene has been recently associated with unipolar major depression [56].
A multifactorial (combination) treatment has been shown to be extremely effective in reducing anxiety and depression in patients with AD [23]. This therapeutic response was APOE- and ACE-dependent. At baseline, all APOE variants showed a similar anxiety and depression rate, except the APOE-4/4 carriers who differed from the rest in significantly lower rates of anxiety and depression (Figure 6). Remarkable changes in anxiety were found among different APOE genotypes (Figure 7). Practically, all APOE variants responded with a significant diminution of anxiogenic symptoms, except patients with the APOE-4/4 genotype who only showed a slight improvement. The best responders were APOE-2/4 > APOE-2/3 > APOE-3/3 > APOE-3/4 carriers (Figure 7). The modest anxiolytic effect seen in APOE-4/4 patients might be due to the very low anxiety rate observed at baseline. In any case, APOE-4/4 carriers are the worst responders, with results similar to those obtained in cognitive performance [16]; however, the potential influence of APOE variants on anxiety and cognition in AD does not show a clear parallelism, suggesting that other more complex mechanisms are involved in the onset of anxiety in dementia. Concerning depression, all APOE genotypes improved their depressive symptoms with treatment except those with the APOE-4/4 genotype which worsen along the treatment period, especially after 9 months (Figure 8). The best responders were APOE-2/4 > APOE-2/3 > APOE-3/3 > APOE-3/4, and the worst responder was APOE-4/4 (Figure 8).
In humans, APOE-4 carriers with deep white matter hyperintesities in Magnetic Resonance Imaging (MRI) show association with depressive symptoms and vascular depression [72]. Reduced caudate nucleus volumes and genetic determinants of homocysteine metabolism accumulate in patients with psychomotor slowing and cognitive deficits [73], and older depressed subjects have persisting cognitive impairments associated with hippocampal volume reduction [74,75]. Depressive symptoms are also associated with stroke and atherogenic lipid profile [76].
During the past two decades, antipsychotic, anti-anxiety and cognitive-enhancing effects have been attributed to ACE inhibitors [77,78]. It has been reported that some ACE inhibitors (captopril, SQ29,852) display similar effects to benzodiazepines in dealing with anxiety-related behaviors in animals [79], and another ACE inhibitor (ceronapril) might shared with neuroleptic drugs an ability to enhance latent inhibition in learning tasks [77]. One SNP (rs4291) located in the promoter region of the ACE gene has been recently associated with unipolar major depression [56].
A multifactorial (combination) treatment has been shown to be extremely effective in reducing anxiety and depression in patients with AD [23]. This therapeutic response was APOE- and ACE-dependent. At baseline, all APOE variants showed a similar anxiety and depression rate, except the APOE-4/4 carriers who differed from the rest in significantly lower rates of anxiety and depression (Figure 6). Remarkable changes in anxiety were found among different APOE genotypes (Figure 7). Practically, all APOE variants responded with a significant diminution of anxiogenic symptoms, except patients with the APOE-4/4 genotype who only showed a slight improvement. The best responders were APOE-2/4 > APOE-2/3 > APOE-3/3 > APOE-3/4 carriers (Figure 7). The modest anxiolytic effect seen in APOE-4/4 patients might be due to the very low anxiety rate observed at baseline. In any case, APOE-4/4 carriers are the worst responders, with results similar to those obtained in cognitive performance [16]; however, the potential influence of APOE variants on anxiety and cognition in AD does not show a clear parallelism, suggesting that other more complex mechanisms are involved in the onset of anxiety in dementia. Concerning depression, all APOE genotypes improved their depressive symptoms with treatment except those with the APOE-4/4 genotype which worsen along the treatment period, especially after 9 months (Figure 8). The best responders were APOE-2/4 > APOE-2/3 > APOE-3/3 > APOE-3/4, and the worst responder was APOE-4/4 (Figure 8).
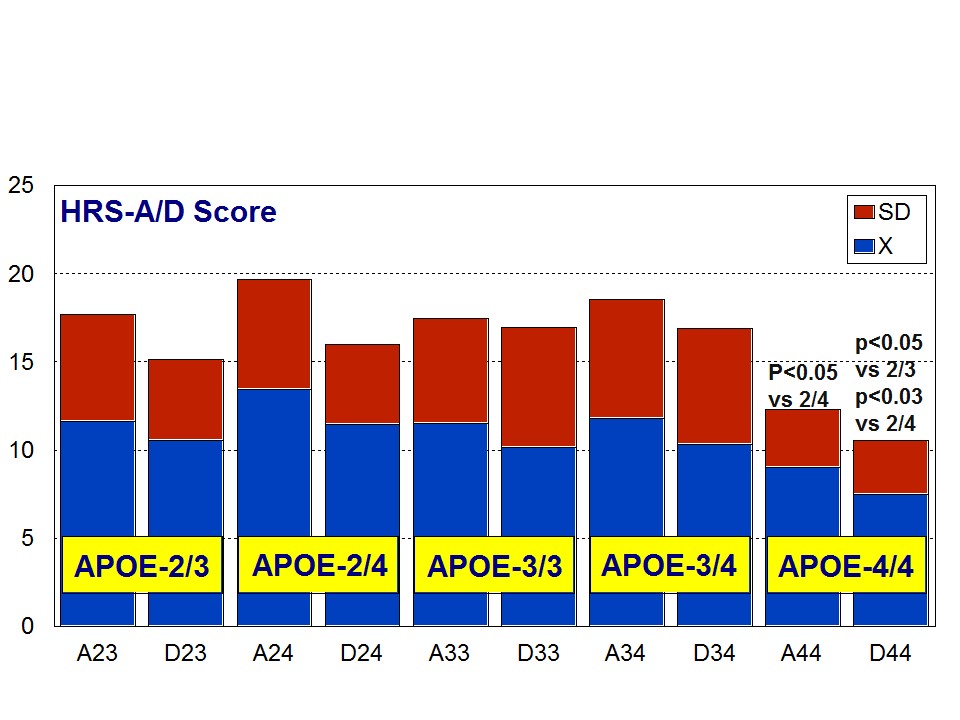 Figure 6: APOE-Related Anxiety (A) and Depression (D) rates (baseline levels) in patients with Alzheimer’s disease.
Figure 6: APOE-Related Anxiety (A) and Depression (D) rates (baseline levels) in patients with Alzheimer’s disease.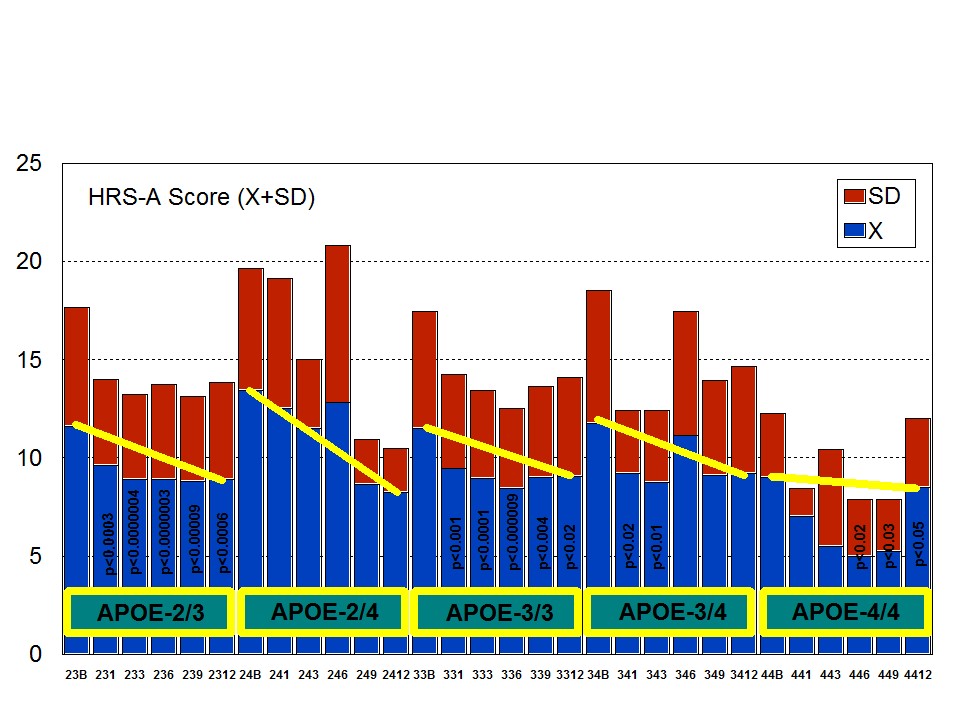 Figure 7: APOE-related anxiety rate in patients with Alzheimer’s disease treated with a combination therapy.
Figure 7: APOE-related anxiety rate in patients with Alzheimer’s disease treated with a combination therapy.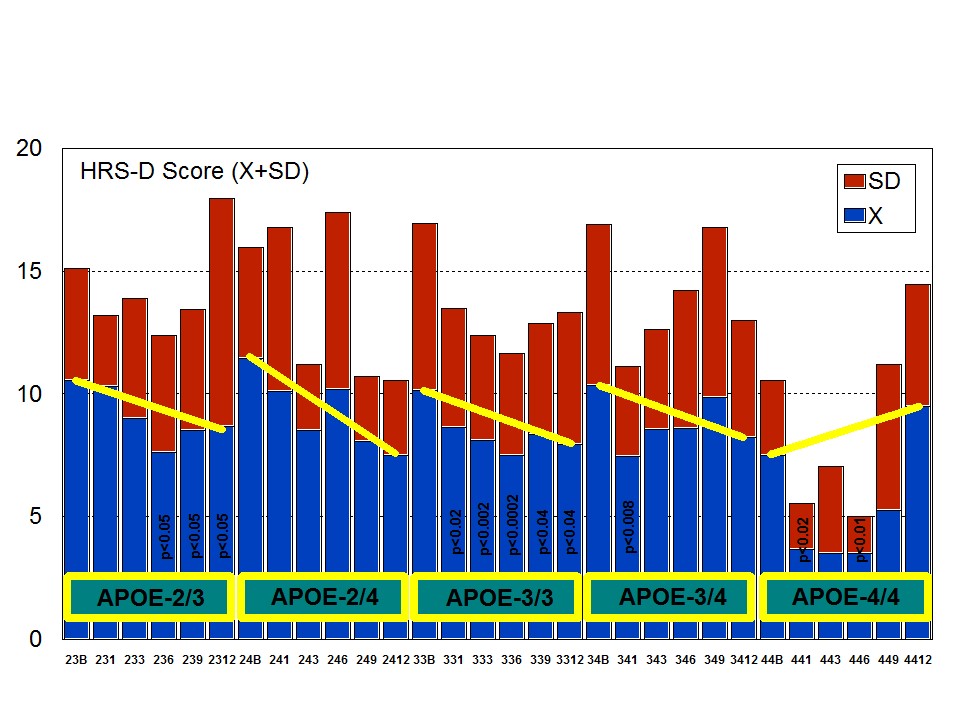 Figure 8: APOE-related depression rate in patients with Alzheimer’s disease treated with a combination therapy.
Figure 8: APOE-related depression rate in patients with Alzheimer’s disease treated with a combination therapy.Patients with each one of the 3 ACE-I/D indel variants are equally anxiogenic (Figure 9) and depressive (Figure 10) at baseline and all of them favourably respond to the multifactorial protocol by gradually reducing anxiety and depressive symptoms along the 12-month treatment period (Figures 9 and 10). The best responders are ACE-I/D followed by ACE-D/D and ACE-I/I, the latter exhibiting the less significant change in anxiogenic parameters (Figure 9); in ACE-D/D carriers the anxiolytic response is faster and more sustainable during the treatment period than in the other genotypes, whereas in ACE-I/D the response is gradual reaching significant values after 9 months of treatment; in contrast, ACE-I/I patients show a very positive response during the first trimester of treatment with an apparent relapse of anxiogenic symptomatology thereafter (Figure 9). This differential ACE-related anxiety pattern might suggest some influence of ACE-I/D variants on mood disorders in AD. Depressive symptoms are also similarly improved in all ACE-I/D variants. The best responders are the heterozygous ACE-I/D followed by the homozygous ACE-D/D and ACE-I/I (Figure 10). Comparatively, the worst responders among ACE-I/D variants are carriers of the ACE-I/I genotype which are also the poorer responders in anxiety and cognition [23].
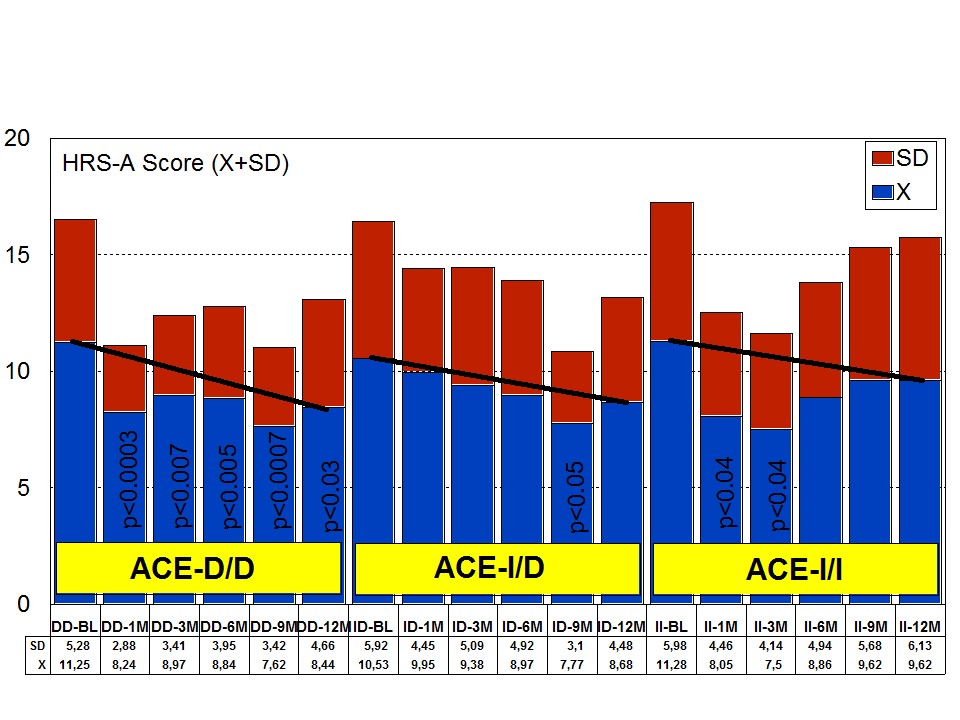 Figure 9: ACE-related anxiety rate in patients with Alzheimer’s disease treated with a combination therapy.
Figure 9: ACE-related anxiety rate in patients with Alzheimer’s disease treated with a combination therapy.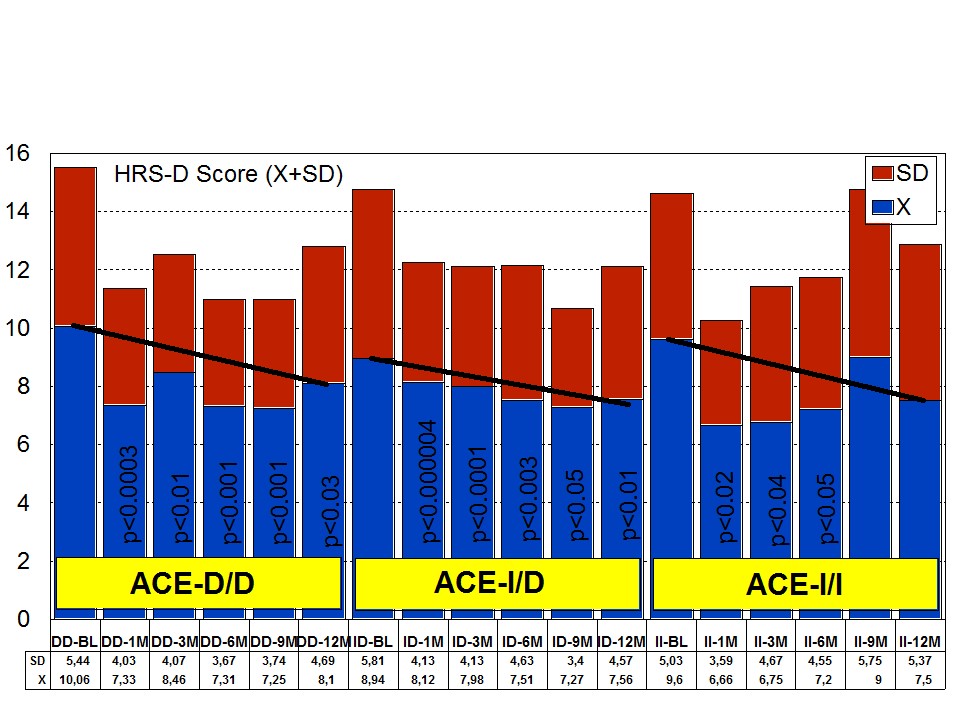 Figure 10: ACE-related depression rate in patients with Alzheimer’s disease treated with a combination therapy.
Figure 10: ACE-related depression rate in patients with Alzheimer’s disease treated with a combination therapy.Effect of APOE-ACE interactions on anxiety and depression
The combination of APOE and ACE polymorphic variants in bigenic clusters yields different anxiety and depression patterns at baseline (Figures 11 and 12) and after one-year treatment (Figures 13 and 14). The most anxiogenic patients at baseline are those with the DD23, ID44, and II34 genotypes, and the less anxiogenic patients are those harbouring the II23, DD44, and ID23 genotypes (Figure 11). The most depressive clusters at baseline are those harbouring the DD23, ID33, and II33 genotypes, with a clear accumulation of APOE-3/3 carriers in these groups, and the less depressive clusters are those represented by carriers of the II23, ID44, and ID23 genotypes (Figure 12). All bigenic clusters show a positive anxiolytic response to the multifactorial protocol, except DD44 which exhibits the worst response by large (Figure 13). The sequence of good anxiogenic responders from better to worse is the following: ID33 > ID44 > DD34 > DD33 > ID34 > II33 > ID23 > II23 = II34 (Figure 13). In a similar fashion, depressive symptoms gradually improved in most bigenic clusters except in DD44 and ID44 subjects in whom the depressive symptomatology tended to deteriorate. The best responders in depression were DD34 > ID33 > DD23 > II23 > ID34 > II34 > ID23 > DD33 > II33, and the worst responders were ID44 and DD44 (Figure 14). As in the case of cognition, DD44 patients represent the poorest responders in anxiety and depression symptoms after one year of treatment, clearly indicating that the association of the APOE-4/4 and ACE-D/D genotypes plays a severe deleterious role on mental performance, at least in cognition and mood [23].
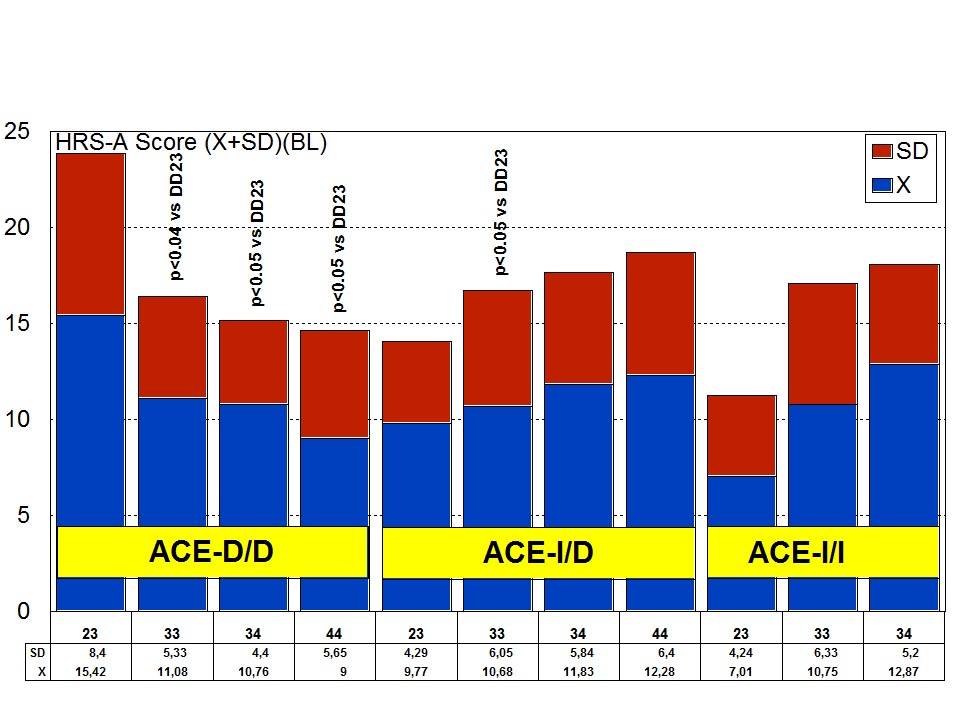 Figure 11: APOE+ACE-related anxiety rate (baseline levels) in patients with Alzheimer’s disease.
Figure 11: APOE+ACE-related anxiety rate (baseline levels) in patients with Alzheimer’s disease.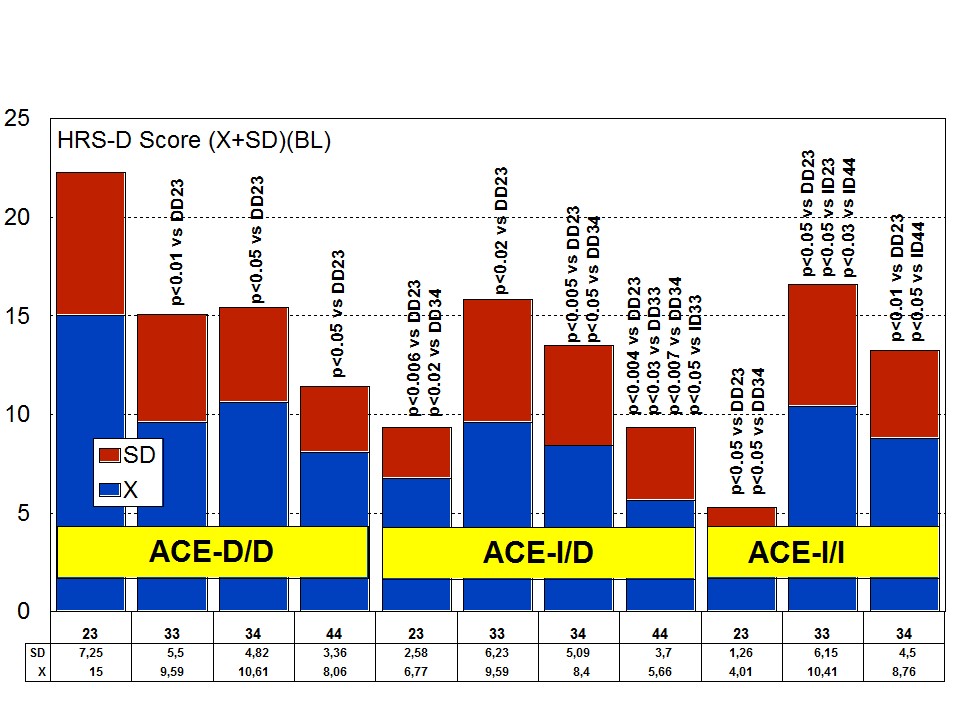 Figure 12: APOE+ACE-related depression rate (baseline levels) in patients with Alzheimer’s disease.
Figure 12: APOE+ACE-related depression rate (baseline levels) in patients with Alzheimer’s disease.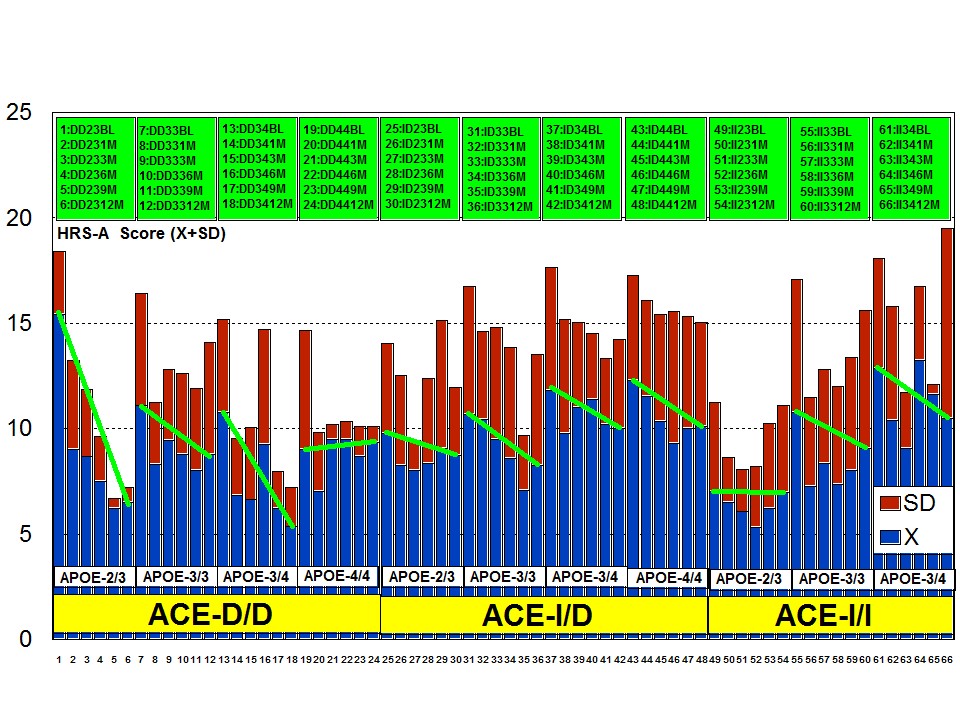
Figure 13: ACE+APOE-related anti-anxiety effect of a multifactorial treatment in patients with Alzheimer’s disease.
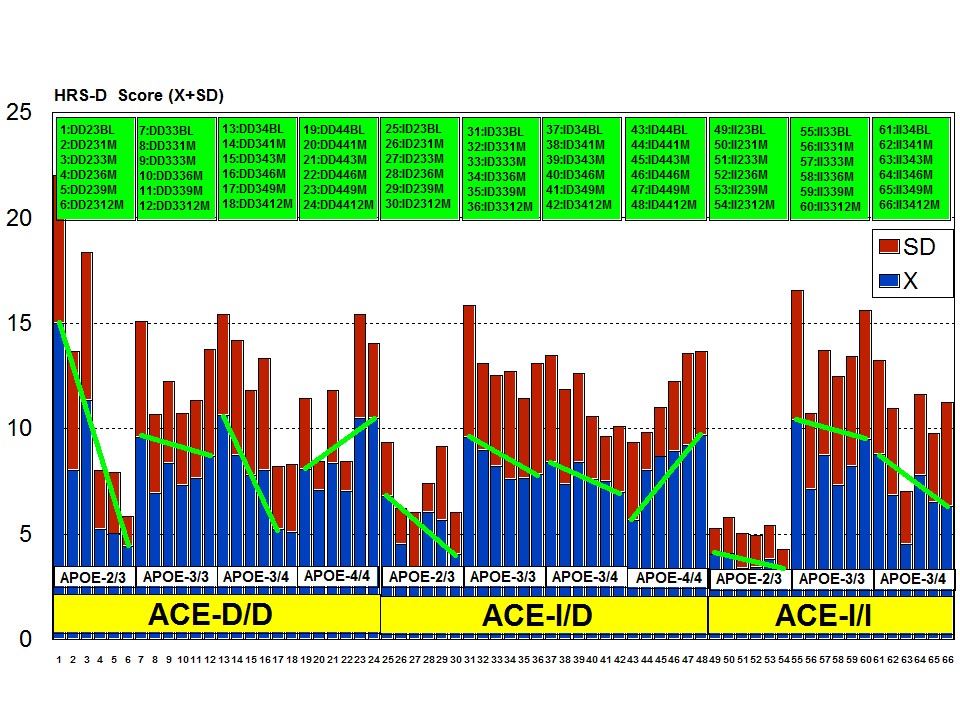
Figure 14: ACE+APOE-related anti-depressant effect of a multifactorial treatment in patients with Alzheimer’s disease.
PHARMACOGENOMICS OF ANTIDEPRESSANTS
Selective Serotonin Reuptake Inhibitors (SSRI) and Selective Serotonin and Norepinephrine Reuptake Inhibitors (SSNRI)
Citalopram: Citalopram is a SSRI which selectively inhibits serotonin reuptake in the presynaptic neurons and has minimal effects on norepinephrine or dopamine. Pathogenic genes potentially associated with citalopram include ABCB1, BDNF, CREB1, CRHR1, CRHR2, FKBP5, GRIA3, GRIK2, GRIK4, GSK3B, HTR1A, HTR1B, HTR2A, MAOA, SLC6A4, TPH1, TPH2. Citalopram-related mechanistic genes are ADRs, CHRMs, DRDs, FKBP5, GABRs, GRIK4, HRHs, HTR1A, HTR1B, HTR1D, HTR2A, SLC6A4 and TPH1. Citalopram is a major substrate of ABCC1, COMT, CYP2C19 and CYP3A4/5, and a minor substrate of CYP2D6. This antidepressant acts as a weak inhibitor of ABCB1, CYP1A2, CYP2B6, CYP2C19, CYP2D6, MAOA, and MAOB; and it is transported by ABCB1 and SLC6A4 [1] (Table 2).
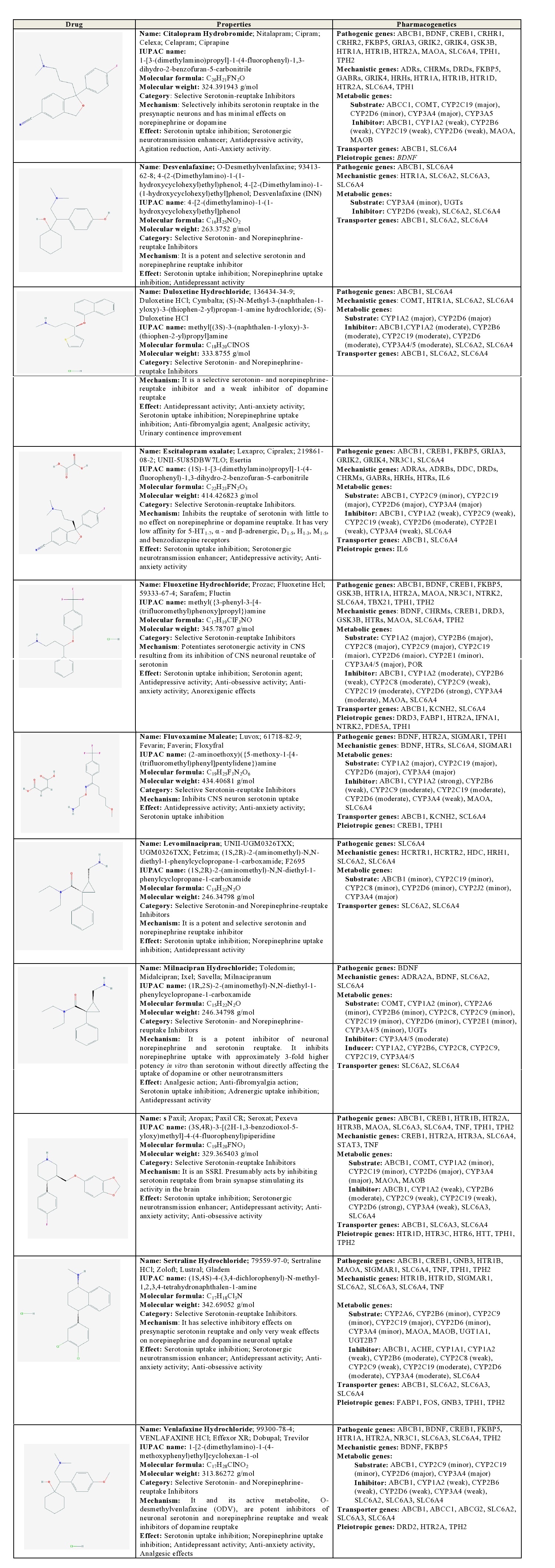
Table 2: Pharmacological profile and pharmacogenetics of Selective Serotonin Reuptake Inhibitors (SSRI) and Selective Serotonin and Norepinephrine Reuptake Inhibitors (SSNRI).
ABCB1: ATP-binding Cassette, sub-family B (MDR/TAP), member 1; ABCC1: ATP-binding Cassette, sub-family C (CFTR/MRP), member 1; ABCG2: ATP-binding Cassette, sub-family G (WHITE), member 2 (Junior blood group); ACHE: Acetylcholinesterase (Yt blood group); ADRA2A: Adrenoceptor Alpha 2A; ADRAs: Adrenoceptors Alpha; ADRBs: Adrenoceptors Beta; ADRs: Adrenoceptors; BDNF: Brain-derived Neurotrophic Factor; CHRMs: Cholinergic Receptors, Muscarinic type; COMT: Catechol-O-methyltransferase; CREB1: cAMP Responsive Element Binding protein 1; CRHR1: Corticotropin Releasing Hormone Receptor 1; CRHR2: Corticotropin Releasing Hormone Receptor 2; CYP1A1: Cytochrome P450, family 1, subfamily A, polypeptide 1; CYP1A2: Cytochrome P450, family 1, subfamily A, polypeptide 2; CYP2A6: Cytochrome P450, family 2, subfamily A, polypeptide 6; CYP2B6: Cytochrome P450, family 2, subfamily B, polypeptide 6; CYP2C8: Cytochrome P450, family 2, subfamily C, polypeptide 8; CYP2C9:Cytochrome P450, family 2, subfamily C, polypeptide 9; CYP2C19: Cytochrome P450, family 2, subfamily C, polypeptide 19; CYP2D6: Cytochrome P450, family 2, subfamily D, polypeptide 6; CYP2E1: Cytochrome P450, family 2, subfamily E, polypeptide 1; CYP2J2: Cytochrome P450, family 2, subfamily J, polypeptide 2; CYP3A4: Cytochrome P450, family 3, subfamily A, polypeptide 4; CYP3A4/5: Cytochrome P450, family 3, subfamily A, polypeptide 4/5; DDC: Dopa decarboxylase (aromatic L-amino acid decarboxylase); DRD2: Dopamine Receptor D2; DRD3: Dopamine Receptor D3; DRDs: Dopamine Receptors; FABP1: Fatty Acid Binding Protein 1, liver; FKBP5: FK506 Binding Protein 5; FOS: FBJ Osteosarcoma oncogene; GABRs: Gamma-aminobutyric acid (GABA) Receptors; GNB3: Guanine Nucleotide binding protein (G protein) Beta polypeptide 3; GRIA3: Glutamate Receptor Ionotropic AMPA 3; GRIK2: Glutamate Receptor Ionotropic Kainate 2; GRIK4: Glutamate Receptor Ionotropic Kainate 4; GSK3B: Glycogen Synthase Kinase 3 Beta; HCRTR1: Hypocretin (orexin) Receptor 1; HCRTR2: Hypocretin (orexin) Receptor 2; HDC: Histidine Decarboxylase; HRH1: Histamine Receptor H1; HRHs: Histamine Receptors; HTR1A: 5-hydroxytryptamine (serotonin) Receptor 1A G protein-coupled; HTR1B: 5-hydroxytryptamine (serotonin) Receptor 1B G protein-coupled; HTR1D: 5-hydroxytryptamine (serotonin) Receptor 1D G protein-coupled; HTR2A: 5-hydroxytryptamine (serotonin) Receptor 2A G protein-coupled; HTR3A: 5-hydroxytryptamine (serotonin) Receptor 3A ionotropic; HTR3B: 5-hydroxytryptamine (serotonin) Receptor 3B ionotropic; HTR3C: 5-hydroxytryptamine (serotonin) Receptor 3C ionotropic; HTR6: 5-hydroxytryptamine (serotonin) Receptor 6 G protein-coupled; HTRs: 5-hydroxytryptamine (serotonin) Receptors; HTT: Huntingtin; IFNA1: Interferon Alpha 1; IL6: Interleukin 6; KCNH2: Potassium Channel, voltage gated eag related subfamily H, member 2; MAOA: Monoamine Oxidase A; NR3C1: Nuclear Receptor subfamily 3 group C member 1 (glucocorticoid receptor); NTRK2: Neurotrophic Tyrosine Kinase Receptor type 2; PDE5A: Phosphodiesterase 5A cGMP-specific; POR: P450 (cytochrome) Oxidoreductase; SCL6A4: Solute Carrier Family 6 (neurotransmitter transporter) member 4; SIGMAR1: Sigma non-opioid intracellular Receptor 1; SLC6A2: Solute Carrier family 6 (neurotransmitter transporter) member 2; SLC6A3: Solute Carrier family 6 (neurotransmitter transporter) member 3; SLC6A4: Solute Carrier family 6 (neurotransmitter transporter) member 4; STAT3: Signal Transducer and Activator of Transcription 3 (acute-phase response factor); TBX21: T-box 21; TNF: Tumor Necrosis Factor; TPH1: Tryptophan Hydroxylase 1; UGT1A1: UDP Glucuronosyltransferase 1 family polypeptide A1; UGT2B7: UDP Glucuronosyltransferase 2 family polypeptide B7; UGTs: UDP Glucuronosyltransferase family.
Desvenlafaxine: Desvenlafaxine is a potent SSNRI, acting as a substrate of CYP3A4 and UGTs, and a weak inhibitor of CYP2D6, SLC6A2, and SLC6A4. ABCB1, SLC6A2, and SLC6A4 proteins are current transporters of venlafaxine [1] (Table 2).
Duloxetine: Duloxetine is a SSNRI and a weak inhibitor of dopamine reuptake, acting as a major substrate of CYP1A2 and CYP2D6, and a moderate inhibitor of ABCB1, CYP1A2, CYP2B6, CYP2C19, CYP2D6, CYP3A4/5, SLC6A2, and SLC6A4. Duloxetine is transported by ABCB1, SLC6A2, and SLC6A4 proteins [1] (Table 2).
Escitalopram: Escitalopram is a SSRI with little effect on norepinephrine or dopamine reuptake, and very low affinity for 5-HT1-7, α - and β-adrenergic, D1-5, H1-3, M1-5, and benzodiazepine receptors. This drug is a major substrate of ABCB1, CYP2C19, CYP2D6, and CYP3A4, and a minor substrate of CYP2C9. Escitalopram is also a moderate inhibitor of ABCB1 and CYP2D6, and a weak inhibitor of CYP1A2, CYP2C9, CYP2C19, CYP2D6, CYP2E1, CYP3A4, and SLC6A4. Its major transporters are ABCB1 and SLC6A4 proteins [1] (Table 2).
Fluoxetine: Fluoxetine is a potent SSRI. Different pathogenic (ABCB1, BDNF, CREB1, FKBP5, GSK3B, HTR1A, HTR2A, MAOA, NR3C1, NTRK2, SLC6A4, TBX21, TPH1, TPH2) and mechanistic genes (BDNF, CHRMs, CREB1, DRD3, GSK3B, HTRs, MAOA, SLC6A4, TPH2) and their products are involved in its therapeutic effect and mechanism of action, respectively. Fluoxetine is a major substrate of CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, and CYP3A4/5, a minor substrate of CYP2E1 and POR, a strong inhibitor of CYP2D6, a moderate inhibitor of CYP1A2 and CYP3A4/5, and a weak inhibitor of ABCB1, CYP2B6, CYP2C9, CYP2C19, MAOA, and SLC6A4. ABCB1, KCNH2, and SLC6A4 proteins are major fluoxetine transporters [1] (Table 2).
Fluvoxamine: Fluvoxamine is a SSRI, a major substrate of CYP1A2, CYP2C19, CYP2D6, and CYP3A4, a strong inhibitor of CYP1A2, a moderate inhibitor of CYP2C9, CYP2C19, and CYP2D6, and a weak inhibitor of ABCB1, CYP2B6, CYP3A4, MAOA, and SLC6A4. Fluvoxamine is transported by ABCB1, KCNH2, and SCL6A4 proteins [1] (Table 2).
Milnacipran: Milnacipran is a potent SSNRI, acting as a minor substrate of COMT, CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1, CYP3A4/5, and UGTs, a moderate inhibitor of CYP3A4/5, and an inducer of CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, and CYP3A4/5. Milnacipran is transported by SLC6A2 and SLC6A4 proteins [1] (Table 2).
Paroxetine: Paroxetine is a SSRI, a major substrate of ABCB1, COMT, CYP2D6, and CYP3A4, a minor substrate of CYP1A2, CYP2C19, MAOA, and MAOB, a strong inhibitor of CYP2D6, a moderate inhibitor of CYP2B6, and a weak inhibitor of ABCB1, CYP1A2, CYP2C9, CYP2C19, CYP3A4, SLC6A3, and SLC6A4. ABCB1, SLC6A3 and SLC6A4 are regular transporters of paroxetine. Some important pathogenic genes (ABCB1, CREB1, HTR1B, HTR2A, HTR3B, MAOA, SLC6A3, SLC6A4, TNF, TPH1, TPH2) might be involved in the efficacy of paroxetine [1] (Table 2).
Sertraline: Sertraline is a SSRI with very weak effects on norepinephrine and dopamine neuronal uptake. Pathogenic (ABCB1, CREB1, GNB3, HTR1B, MAOA, SIGMAR1, SLC6A4, TNF, TPH1, TPH2) and mechanistic genes (HTR1B, HTR1D, SIGMAR1, SLC6A2, SLC6A3, SLC6A4, TNF) are involved in the antidepressant effect of sertraline, which is a major substrate of CYP2C19, a minor substrate of CYP2A6, CYP2B6, CYP2C9, CYP2D6, CYP3A4, MAOA, MAOB, UGT1A1, and UGT2B7, a moderate inhibitor of CYP2B6, CYP2C19, CYP2D6, and CYP3A4, and a weak inhibitor of ABCB1, ACHE, CYP1A1, CYP1A2, CYP2C8, CYP2C9, and SLC6A4. Its principal transporters are ABCB1, SLC6A2, SLC6A3, and SLC6A4 proteins [1] (Table 2).
Venlafaxine: Venlafaxine is a SSNRI and a weak inhibitor of dopamine reuptake, with potential, reciprocal influence on some pathogenic genes (ABCB1, BDNF, CREB1, FKBP5, HTR1A, HTR2A, NR3C1, SLC6A3, SLC6A4, TPH2). Venlafaxine is a major substrate of CYP2D6 and CYP3A4 enzymes, a minor substrate of ABCB1, CYP2C9, and CYP2C19, and a weak inhibitor of ABCB1, CYP1A2, CYP2B6, CYP2D6, CYP3A4, SLC6A2, SLC6A3, and SLC6A4. Several genes (ABCB1, ABCC1, ABCG2, SLC6A2, SLC6A3, SLC6A4) encode transporter proteins for the penetration of venlafaxine into the brain [1] (Table 2).
Tricyclics (TCAs) and other norepinephrine-reuptake inhibitors
Amitriptyline: Amitriptyline is a tricyclic drug which increases the synaptic concentration of serotonin and/or norepinephrine in the CNS by inhibiting their reuptake at the presynaptic neuronal membrane. Pathogenic and mechanistic genes are associated with the therapeutic effects of amitriptyline. It is a major substrate of ABCB1, CYP2D6, CYP3A4/5, GSTP1, UGT1A3, UGT1A4, and UGT2B10, a minor substrate of CYP1A2, CYP2B6, CYP2C9, and CYP2C19, a moderate inhibitor of ABCB1, ABCC2, ABCG2, CYP1A2, CYP2C9, CYP2C19, and CYP2D6, and a weak inhibitor of CYP2E1. ABCB1, ABCC2, ABCG2, KCNE2, KCNH2, KCNQ1, SCN5A, and SLC6A4 proteins are current transporters of amitriptyline [1] (Table 3).
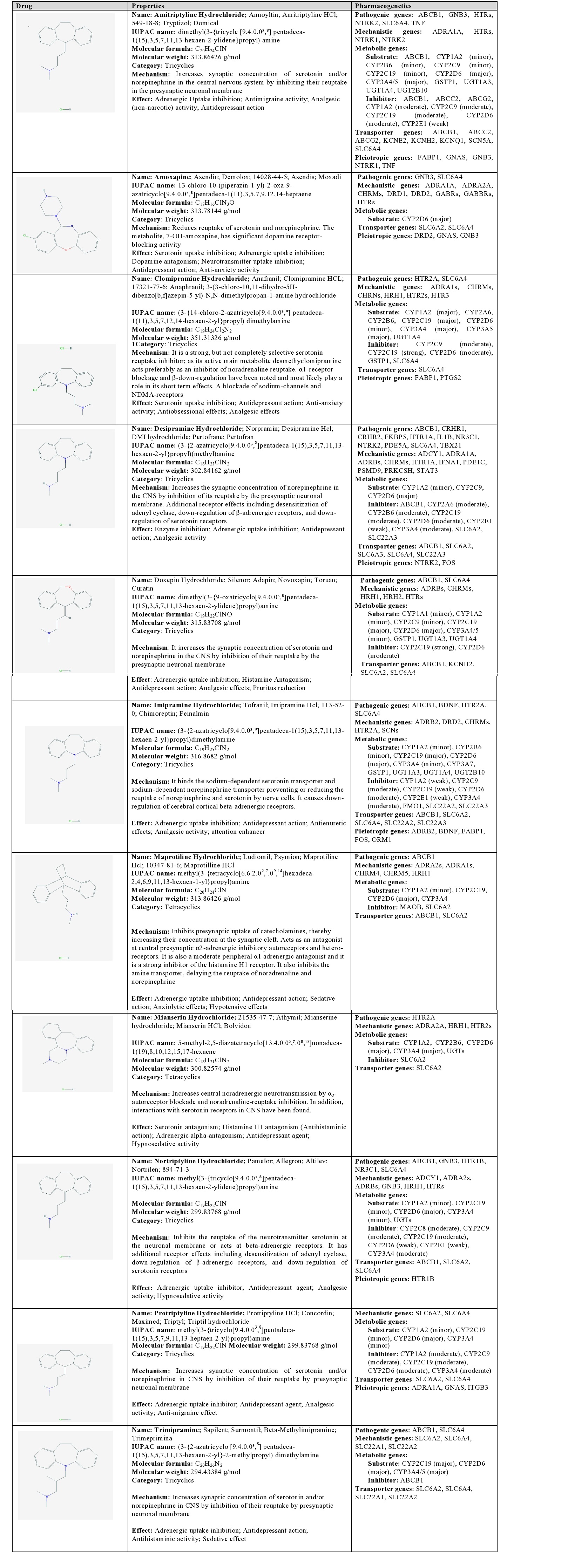
Table 3: Pharmacological profile and pharmacogenetics of Tricyclics (TCAs) and other norepinephrine-reuptake inhibitors.
ABCB1: ATP-binding Cassette, sub-family B (MDR/TAP) member 1; ABCC2: ATP-binding Cassette sub-family C (CFTR/MRP) member 2; ABCG2: ATP-binding Cassette sub-family G (WHITE) member 2 (Junior blood group); ADCY1: Adenylate Cyclase 1 (brain); ADRA1A: Adrenoceptor Alpha 1A; ADRA1s: Adrenoceptors alpha 1; ADRA2A: Adrenoceptor Alpha 2A; ADRA2s: Adrenoceptors Alpha 2; ADRB2: Adrenoceptor Beta 2 surface; ADRBs: Adrenoceptors Beta; BDNF: Brain-derived Neurotrophic Factor; CHRM4: Cholinergic Receptor Muscarinic 4; CHRM5: Cholinergic Receptor Muscarinic 5; CHRMs: Cholinergic Receptors Muscarinic type; CHRNs: Cholinergic Receptors Nicotinic type; CRHR1: Corticotropin Releasing Hormone Receptor 1; CRHR2: Corticotropin Releasing Hormone Receptor 2; CYP1A1: Cytochrome P450 family 1 subfamily A polypeptide 1; CYP1A2: Cytochrome P450 family 1 subfamily A polypeptide 2; CYP2A6: Cytochrome P450 family 2 subfamily A polypeptide 6; CYP2B6 Cytochrome P450 family 2 subfamily B polypeptide 6; CYP2C19: Cytochrome P450 family 2 subfamily C polypeptide 19; CYP2C8: Cytochrome P450 family 2 subfamily C polypeptide 8; CYP2C9: Cytochrome P450 family 2 subfamily C polypeptide 9; CYP2D6: Cytochrome P450 family 2 subfamily D polypeptide 6; CYP2E1: Cytochrome P450 family 2 subfamily E polypeptide 1; CYP3A4: Cytochrome P450 family 3 subfamily A polypeptide 4; CYP3A4/5: Cytochrome P450 family 3 subfamily A polypeptide 4/5; CYP3A5: Cytochrome P450 family 3 subfamily A polypeptide 5; CYP3A7: Cytochrome P450 family 3 subfamily A polypeptide 7; DRD1: Dopamine Receptor D1; DRD2: Dopamine Receptor D2; FABP1: Fatty Acid Binding Protein 1 liver; FKBP5: Binding Protein 5; FMO1: Flavin containing Monooxygenase 1; FOS: FBJ murine Osteosarcoma viral oncogene homolog; GABBRs: Gamma-aminobutyric acid (GABA) A receptors Beta; GABRs: Gamma-aminobutyric acid (GABA) A Receptors; GNAS: GNAS complex locus; GNB3: Guanine Nucleotide binding protein (G protein) Beta polypeptide 3; GSTP1: Glutathione S-transferase Pi 1; HRH1: Histamine Receptor H1; HRH2: Histamine Receptor H2; HTR1A: 5-hydroxytryptamine (serotonin) Receptor 1A G protein-coupled; HTR1B: 5-hydroxytryptamine (serotonin) Receptor 1B G protein-coupled; HTR2A: 5-hydroxytryptamine (serotonin) Receptor 2A G protein-coupled; HTR2s: 5-hydroxytryptamine (serotonin) Receptors 2; HTR3: Histone H3; HTRs: 5-hydroxytryptamine (serotonin) Receptors; IFNA1: Interferon Alpha 1; IL1B: Interleukin 1 Beta; ITGB3: Integrin Beta 3 (platelet glycoprotein IIIa, antigen CD61); KCNE2: Potassium Channel voltage gated subfamily E regulatory beta subunit 2; KCNH2: Potassium Channel voltage gated eag related subfamily H member 2; KCNQ1: Potassium Channel voltage gated KQT-like subfamily Q member 1; MAOB: Monoamine Oxidase B; NR3C1: Nuclear Receptor subfamily 3 group C member 1 (glucocorticoid receptor); NTRK1: Neurotrophic Tyrosine Kinase Receptor type 1; NTRK2 Neurotrophic Tyrosine Kinase Receptor type 2; ORM1: Orosomucoid 1; PDE1C: Phosphodiesterase 1C, calmodulin-dependent 70kDa; PDE5A: Phosphodiesterase 5A, cGMP-specific; PRKCSH: Protein Kinase C Substrate 80K-H; PSMD9: Proteasome (prosome, macropain) 26S subunit non-ATPase 9; PTGS2: Prostaglandin-endoperoxide Synthase 2 (prostaglandin G/H synthase and cyclooxygenase); SCN5A: Sodium Channel voltage gated type V alpha subunit; SCNs: Sodium Channels voltage gated; SLC22A1: Solute Carrier family 22 (organic cation transporter) member 1; SLC22A2: Solute Carrier family 22 (organic cation transporter) member 2; SLC22A3: Solute Carrier family 22 (organic cation transporter) member 3; SLC6A2: Solute Carrier family 6 (neurotransmitter transporter) member 2; SLC6A3: Solute Carrier family 6 (neurotransmitter transporter) member 3; SLC6A4: Solute Carrier family 6 (neurotransmitter transporter) member 4; STAT3: Signal Transducer and Activator of Transcription 3 (acute-phase response factor); TBX21: T-box 21; TNF: Tumor Necrosis Factor; UGT1A3: UDP Glucuronosyltransferase 1 family polypeptide A3; UGT1A4: UDP Glucuronosyltransferase 1 family polypeptide A4; UGT2B10: UDP Glucuronosyltransferase 2 family polypeptide B10; UGTs: UDP Glucuronosyltransferase family.
Amoxapine: Amoxapine is a tricyclic drug which reduces the reuptake of serotonin and norepinephrine at the synaptic cleft. Its metabolite 7-OH-amoxapine has a significant dopamine receptor-blocking activity. Amoxapine is a major substrate of CYP2D6 enzymes and is transported by SLC6A2 and SLC6A4 [1] (Table 3).
Clomipramine: Clomipramine is a strong, non-selective serotonin reuptake inhibitor. Its main metabolite desmethylclomipramine acts as an inhibitor of noradrenaline reuptake. This tricyclic also displays α1-receptor blocking activity, β-down-regulation, and blockade of sodium-channels and NMDA receptors. Clomipramine is a major substrate of CYP1A2, CYP2A6, CYP2B6, CYP2C19, CYP3A4/5, and UGT1A4, a minor substrate of CYP2D6, a strong inhibitor of CYP2C19, and a moderate inhibitor of CYP2C9, CYP2D6, GSTP1, and SLC6A4. SLC6A4 is its main transporter [1] (Table 3).
Desipramine: Desipramine is a tricyclic drug which increases the synaptic concentration of norepinephrine in the CNS by inhibition of its reuptake at the presynaptic neuronal membrane. Additional receptor effects include desensitization of adenyl cyclase, down-regulation of β-adrenergic receptors, and down-regulation of serotonin receptors. Diverse pathogenic (ABCB1, CRHR1, CRHR2, FKBP5, HTR1A, IL1B, NR3C1, NTRK2, PDE5A, SLC6A4, TBX21) and mechanistic genes (ADCY1, ADRA1A, ADRBs, CHRMs, HTR1A, IFNA1, PDE1C, PSMD9, PRKCSH, STAT3) influence the effects of desipramine. This antidepressant is a major substrate of CYP2D6, and intermediate substrate of CYP2C9, and a minor substrate of CYP1A2, a moderate inhibitor of ABCB1, CYP2A6, CYP2B6, CYP2C19, CYP2D6, CYP3A4, SLC6A2, and SLC22A3, and a weak inhibitor of CYP2E1. Its principal transporters are ABCB1, SLC6A2, SLC6A3, SLC6A4, and SLC22A3 [1] (Table 3).
Doxepin: Doxepin increases the synaptic concentration of serotonin and norepinephrine in the CNS by inhibition of their reuptake at the presynaptic neuronal membrane. This tricyclic is a major substrate of CYP2C19, CYP2D6, GSTP1, UGT1A3, and UGT1A4, a minor substrate of CYP1A1, CYP1A2, CYP2C9, and CYP3A4/5, a moderate inhibitor of CYP2D6, and a strong inhibitor of CYP2C19 enzymes. ABCB1, KCNH2, SLC6A2, and SLC6A4 are its current transporters [1] (Table 3).
Imipramine: Imipramine is a tricyclic drug that binds the sodium-dependent serotonin transporter and sodium-dependent norepinephrine transporter preventing or reducing the reuptake of norepinephrine and serotonin by nerve cells. It also causes down-regulation of cerebral cortical beta-adrenergic receptors. Pathogenic genes (ABCB1, BDNF, HTR2A, and SLC6A4) and mechanistic genes associated with adrenergic, dopaminergic, cholinergic, serotonergic, and histaminergic neurotransmission (ADRB2, DRD2, CHRMs, HTR2A, and SCNs) may affect its pharmacodynamics properties. Imipramine is a major substrate of CYP2C19, CYP2D6, GSTP1, UGT1A3, UGT1A4, and UGT2B10, a minor substrate of CYP1A2, CYP2B6, CYP3A4, and CYP3A7, a weak inhibitor of CYP1A2, CYP2C19, and CYP2E1, and a moderate inhibitor of CYP2C9, CYP2D6, CYP3A4, FMO1, SLC22A2, and SLC22A3. Imipramine is transported by ABCB1, SLC6A2, SLC6A4, SLC22A2, and SLC22A3 proteins [1] (Table 3).
Maprotiline: Maprotiline is a pleiotropic tetracyclic that inhibits the presynaptic uptake of catecholamines, thereby increasing their concentration at the synaptic cleft. Maprotiline also acts as an antagonist at central presynaptic α2-adrenergic inhibitory autoreceptors and hetero-receptors, is a moderate peripheral α1 adrenergic antagonist and is a strong inhibitor of the histamine H1 receptor. It also inhibits the amine transporter, delaying the reuptake of noradrenaline and norepinephrine. Maprotiline is a major substrate of CYP2D6, a minor substrate of CYP1A2, CYP2C19, and CYP3A4 enzymes, inhibits MAOB and SLC6A2, and is transported by ABCB1 and SLC6A2 [1] (Table 3).
Mianserin: Mianserin is a tetracyclic drug that increases central noradrenergic neurotransmission by α2-autoreceptor blockade and noradrenaline-reuptake inhibition, and also interacts with serotonin receptors. This antidepressant is a major substrate of CYP2D6 and CYP3A4/5 enzymes, a minor substrate of CYP1A2, CYP2B6, and UGTs, and an inhibitor of SLC6A2, which is also its main transporter [1] (Table 3).
Nortriptyline: Nortriptyline is a tricyclic antidepressant that inhibits the reuptake of serotonin at the neuronal membrane and also interacts with beta-adrenergic receptors. Other mechanistic effects of nortriptyline include desensitization of adenyl cyclase, down-regulation of β-adrenergic receptors, and down-regulation of serotonin receptors. It is a major substrate of CYP2D6 and UGTs, a minor substrate of CYP1A2, CYP2C19, and CYP3A4, a moderate inhibitor of CYP2C8, CYP2C9, CYP2C19, and CYP3A4, and a weak inhibitor of CYP2D6 and CYP2E1 enzymes. ABCB1, SLC6A2, and SLC6A4 are its principal transporters [1] (Table 3).
Protriptyline: Protriptyline is a tricyclic compound that increases the intersynaptic concentration of serotonin and/or norepinephrine by inhibition of their reuptake at the presynaptic neuronal membrane. It is a major substrate of CYP2D6, a minor substrate of CYP1A2, CYP2C19, and CYP3A4, and a moderate inhibitor of CYP1A2, CYP2C9, CYP2C19, CYP2D6, and CYP3A4. Protriptyline is transported by SLC6A2 and SLC6A4 proteins [1] (Table 3).
Trimipramine: Trimipramine is a tricyclic antidepressant that enhances the concentration of serotonin and/or norepinephrine by inhibition of their reuptake at the presynaptic neuronal membrane. It is a major substrate of CYP2C19, CYP2D6, and CYP3A4/5, inhibits ABCB1, and is transported by SLC6A2, SLC6A4, SLC22A1, and SLC22A2 proteins [1] (Table 3).
Monoamine Oxidase Inhibitors (MAOIs)
Isocarboxacid: Isocarboxacid is a non-selective MAOI that increases endogenous concentrations of epinephrine, norepinephrine, dopamine, and serotonin through inhibition of MAOA and MAOB enzymes. It is a substrate of COMT and CYP2D6, and a strong inhibitor of MAOs [1] (Table 4).
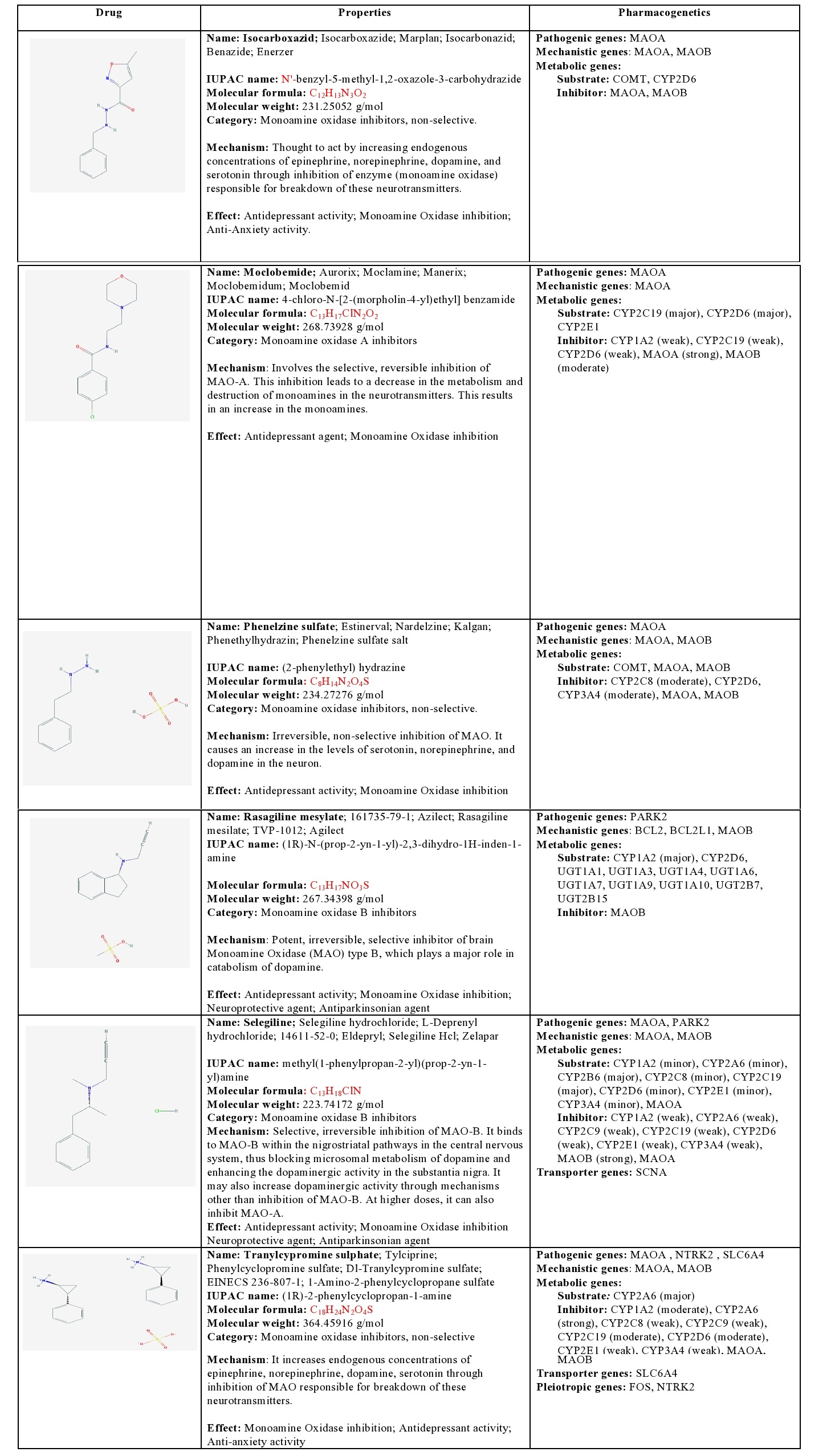
Table 4: Pharmacological profile and pharmacogenetics of Monoamine Oxidase Inhibitors (MAOIs).
BCL2: B-cell CLL/Lymphoma 2; BCL2L1: BCL2-like 1; COMT: Catechol-O-methyltransferase; CYP1A2: Cytochrome P450 family 1 subfamily A polypeptide 2; CYP2A6: Cytochrome P450 family 2 subfamily A polypeptide 6; CYP2B6: Cytochrome P450 family 2 subfamily B polypeptide 6; CYP2C19: Cytochrome P450 family 2 subfamily C polypeptide 19; CYP2C8: Cytochrome P450 family 2 subfamily C polypeptide 8; CYP2C9: Cytochrome P450 family 2 subfamily C polypeptide 9; CYP2D6: Cytochrome P450 family 2 subfamily D polypeptide 6; CYP2E1: Cytochrome P450 family 2 subfamily E polypeptide 1; CYP3A4: Cytochrome P450 family 3 subfamily A polypeptide 4; FOS: FBJ Osteosarcoma oncogene; MAOA: Monoamine Oxidase A; MAOB: Monoamine Oxidase B; NTRK2: Neurotrophic Tyrosine Kinase Receptor type 2; PARK2: Parkin RBR E3 ubiquitin protein ligase; SCNA: Synuclein Alpha (non A4 component of amyloid precursor); SLC6A4: Solute Carrier family 6 (neurotransmitter transporter) member 4; UGT1A1: UDP Glucuronosyltransferase 1 family polypeptide A1; UGT1A10: UDP Glucuronosyltransferase 1 family polypeptide A10; UGT1A3: UDP Glucuronosyltransferase 1 family polypeptide A3; UGT1A4: UDP Glucuronosyltransferase 1 family polypeptide A4; UGT1A6: UDP Glucuronosyltransferase 1 family polypeptide A6; UGT1A7: UDP Glucuronosyltransferase 1 family polypeptide A7; UGT1A9: UDP Glucuronosyltransferase 1 family polypeptide A9; UGT2B15: UDP Glucuronosyltransferase 2 family polypeptide B15; UGT2B7: UDP Glucuronosyltransferase 2 family polypeptide B7.
Moclobemide: Moclobemide is a MAOA inhibitor that inhibits deamination of serotonin, norepinephrine, and dopamine, leading to increased concentrations of these neurotransmitters in the CNS. It is a major substrate of CYP2C19 and CYP2D6, a minor substrate of CYP2E1, a weak inhibitor of CYP1A2, CYP2C19, and CYP2D6, a strong inhibitor of MAOA, and a moderate inhibitor of MAOB [1] (Table 4).
Phenelzine: Phenelzine is a non-selective MAOI that increases endogenous concentrations of norepinephrine, dopamine, and serotonin through inhibition of MAOs. It is a substrate of COMT, MAOA, and MAOB, and a moderate inhibitor of CYP2C8, CYP2D6, CYP3A4, MAOA and MAOB [1] (Table 4).
Rasagiline: Rasagiline is a potent, irreversible, selective inhibitor of brain MAOB. It is a major substrate of CYP1A2, CYP2D6, UGT1A1, UGT1A3, UGT1A4, UGT1A6, UGT1A7, UGT1A9, UGT1A10, UGT2B7, and GT2B15, and a strong inhibitor of MAOB [1] (Table 4).
Selegiline: Selegiline is a selective, irreversible MAOB inhibitor that binds to MAOB within the nigrostriatal pathway, thus blocking microsomal metabolism of dopamine and enhancing the dopaminergic activity in the substantia nigra. At high doses, it can also inhibit MAOA. Selegiline is a major substrate of CYP2B6 and CYP2C19, a minor substrate of CYP1A2, CYP2A6, CYP2C8, CYP2D6, CYP2E1, and CYP3A4, a weak inhibitor of CYP1A2, CYP2A6, CYP2C9, CYP2C19, CYP2D6, CYP2E1, CYP3A4, and MAOA, and a strong inhibitor of MAOB. SCNA is involved in its transport into the brain [1] (Table 4).
Tranylcypromine: Tranylcypromine is a non-selective MAOI that increases endogenous concentrations of epinephrine, norepinephrine, dopamine, and serotonin through inhibition of MAOs. It is a major substrate of CYP2A6, a weak inhibitor of CYP2C8, CYP2E1, and CYP3A4, a moderate inhibitor of CYP1A2, CYP2C19, and a strong inhibitor of CYP2A6 and MAOs. It is transported by SLC6A4 [1] (Table 4).
Other categories of antidepressants
Agomelatine: Agomelatine is a melatonergic agonist and a 5-HT2C antagonist. It is a major substrate of CYP1A1 and CYP1A2, and a minor substrate of CYP2C9 and CYP2C19 [1] (Table 5).
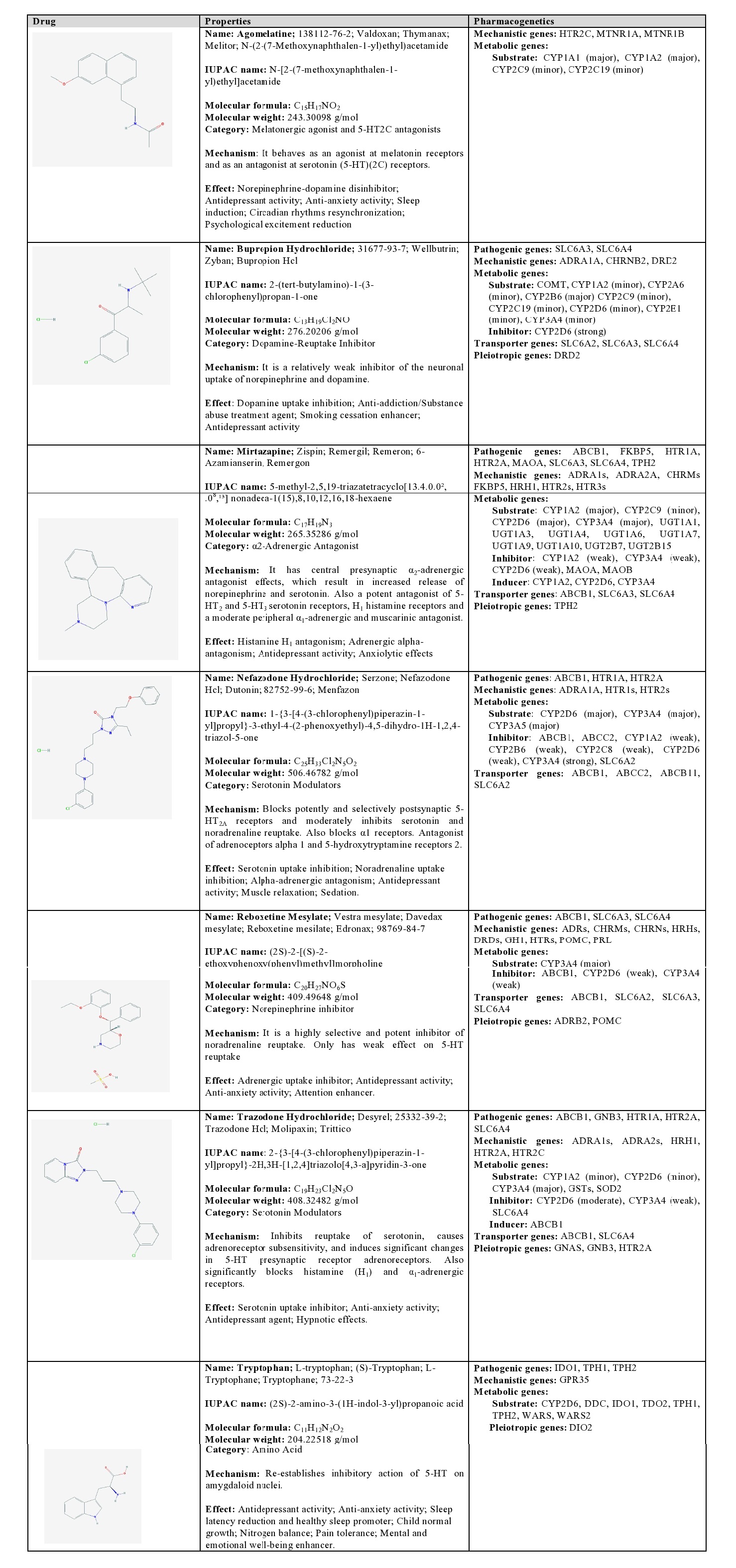 Table 5: Pharmacological profile and pharmacogenetics of other categories of antidepressants.
Table 5: Pharmacological profile and pharmacogenetics of other categories of antidepressants.ABCB1: ATP-binding Cassette sub-family B (MDR/TAP) member 1; ABCB11: ATP-binding Cassette, sub-family B (MDR/TAP) member 11; ABCC2: ATP-binding Cassette sub-family C (CFTR/MRP) member 2; ADRA1A: Adrenoceptor Alpha 1A; ADRA1s: Adrenoceptors Alpha 1; ADRA2A: Adrenoceptor Alpha 2A; ADRA2s: Adrenoceptors Alpha 2; ADRB2: Adrenoceptor Beta 2 surface; ADRs: Adrenoceptors; CHRMs: Cholinergic Receptors Muscarinic type; CHRNB2: Cholinergic Receptor Nicotinic Beta 2 (neuronal); CHRNs: Cholinergic Receptors Nicotinic type; COMT: Catechol-O-methyltransferase; CYP1A1: Cytochrome P450, family 1 subfamily A polypeptide 1; CYP1A2: Cytochrome P450 family 1 subfamily A polypeptide 2; CYP2A6: Cytochrome P450 family 2 subfamily A polypeptide 6; CYP2B6: Cytochrome P450 family 2 subfamily B polypeptide 6; CYP2C19: Cytochrome P450 family 2 subfamily C polypeptide 19; CYP2C8: Cytochrome P450 family 2 subfamily C polypeptide 8; CYP2C9: Cytochrome P450 family 2 subfamily C polypeptide 9; CYP2D6: Cytochrome P450 family 2 subfamily D polypeptide 6; CYP2E1: Cytochrome P450 family 2 subfamily E polypeptide 1; CYP3A4: Cytochrome P450 family 3 subfamily A polypeptide 4; CYP3A5: Cytochrome P450 family 3 subfamily A polypeptide 5; DDC: Dopa Decarboxylase (aromatic L-amino acid decarboxylase); DIO2: Deiodinase Iodothyronine type II; DRD2: Dopamine Receptor D2; DRDs: Dopamine Receptors; FKBP5: FK506 binding protein 5; GH1: Growth Hormone 1; GNAS: GNAS complex locus; GNB3: Guanine Nucleotide binding protein (G protein) Beta polypeptide 3; GPR35: G Protein-coupled Receptor 35; GSTs: Glutathione S-transferases; HRH1: Histamine Receptor H1; HRHs: Histamine Receptors; HTR1A: 5-hydroxytryptamine (serotonin) Receptor 1A G protein-coupled; HTR1s: 5-hydroxytryptamine (serotonin) Receptors 1; HTR2A: 5-hydroxytryptamine (serotonin) Receptor 2A G protein-coupled; HTR2C: 5-hydroxytryptamine (serotonin) Receptor 2C G protein-coupled; HTR2s: 5-hydroxytryptamine (serotonin) Receptors 2; HTR3s: 5-hydroxytryptamine (serotonin) Receptors 3; IDO1: Indoleamine 2,3-dioxygenase 1; MAOA: Monoamine Oxidase A; MAOB: Monoamine Oxidase B; MTNR1A: Melatonin Receptor 1A; MTNR1B: Melatonin Receptor 1B; POMC: Proopiomelanocortin; PRL: Prolactin; SLC6A2: Solute Carrier family 6 (neurotransmitter transporter) member 2; SLC6A3: Solute Carrier family 6 (neurotransmitter transporter) member 3; SLC6A4: Solute Carrier family 6 (neurotransmitter transporter) member 4; SOD2: Superoxide Dismutase 2 mitocondrial; TDO2: Tryptophan 2,3-dioxygenase; TPH1: Tryptophan Hydroxylase 1; TPH2: Tryptophan Hydroxylase 2; UGT1A: UDP Glucuronosyltransferase 1 family polypeptide A complex locus; UGT1A3: UDP Glucuronosyltransferase 1 family polypeptide A3; UGT1A4: UDP Glucuronosyltransferase 1 family polypeptide A4; UGT1A6: UDP Glucuronosyltransferase 1 family polypeptide A6; UGT1A7: UDP Glucuronosyltransferase 1 family polypeptide A7; UGT1A9: UDP Glucuronosyltransferase 1 family polypeptide A9; UGT1A10: UDP Glucuronosyltransferase 1 family polypeptide A10; UGT2B7: UDP Glucuronosyltransferase 2 family polypeptide B7; UGT2B15: UDP Glucuronosyltransferase 2 family polypeptide B15; WARS: Tryptophanyl-tRNA Synthetase; WARS2: Tryptophanyl tRNA Synthetase 2 mitochondrial.
Bupropion: Bupropion is a dopamine-reuptake inhibitor. It is a substrate of COMT, a major substrate of CYP2B6, a minor substrate of CYP1A2, CYP2A6, CYP2C9, CYP2C19, CYP2D6, CYP2E1 and CYP3A4, and a strong inhibitor of CYP2D6. Its transport is modulated by SLC6A2, SLC6A3, and SLC6A4 [1] (Table 5).
Mirtazapine: Mirtazapine is a α2-adrenergic antagonist with pleiotropic effects on other neurotransmitters. It has central presynaptic α2-adrenergic antagonist effects, which result in increased release of norepinephrine and serotonin, and is also a potent antagonist of 5-HT2 and 5-HT3 serotonin receptors, H1 histamine receptors and a moderate antagonist of peripheral α1-adrenergic and muscarinic receptors. Mirtazapine is a major substrate of CYP1A2, CYP2D6, CYP3A4, UGT1A1, UGT1A3, UGT1A4, UGT1A6, UGT1A7, UGT1A9, UGT1A10, UGT2B7, and UGT2B15, a minor substrate of CYP2C9, a weak inhibitor of CYP1A2, CYP3A4, CYP2D6, MAOA and MAOB, and an inducer of CYP1A2, CYP2D6 and CYP3A4. Several pathogenic (ABCB1, FKBP5, HTR1A, HTR2A, MAOA, SLC6A3, SLC6A4, TPH2) and mechanistic genes (ADRA1s, ADRA2A, FKBP5, HRH1, HTRs) are involved in its pharmacodynamic properties, and is transported by ABCB1, SLC6A3 and SLC6A4 [1] (Table 5).
Nefazodone: Nefazodone is a serotonin modulator that potently and selectively blocks postsynaptic 5-HT2A receptors and moderately inhibits serotonin and noradrenaline reuptake. It is an antagonist of alpha-1 adrenoceptors and 5-hydroxytryptamine receptor 2. This compound is a major substrate of CYP2D6, CYP3A4 and CYP3A5, a strong inhibitor of CYP3A4, a moderate inhibitor of ABCB1 and ABCC2, and a weak inhibitor of CYP1A2, CYP2B6, CYP2C8, CYP2D6, and SLC6A2. ABCB1, ABCC2, ABCB11, and SLC6A2 participate in its transport [1] (Table 5).
Reboxetine: Reboxetine is a highly selective and potent inhibitor of noradrenaline reuptake, with weak effects on 5-HT reuptake. It is a major substrate of CYP3A4, and a weak inhibitor of ABCB1, CYP2D6 and CYP3A4. ABCB1, SLC6A2, SLC6A3 and SLC6A4 are its preferential transporters [1] (Table 5).
Trazodone: Trazodone is a pleiotropic serotonin modulator that inhibits the reuptake of serotonin, causes adrenoreceptor subsensitivity, and induces significant changes in 5-HT presynaptic receptors, also blocking histamine (H1) and α1-adrenergic receptors. It is a major substrate of CYP3A4, a minor substrate of CYP1A2, CYP2D6, GSTs, and SOD2, a moderate inhibitor of CYP2D6, a weak inhibitor of CYP3A4 and SLC6A4, and an inducer of ABCB1. Its transport is mediated by ABCB1 and SLC6A4 transporters [1] (Table 5).
COMPARATIVE STUDIES
According to Ereshefsky et al., [80] the Selective Serotonin Reuptake Inhibitors (SSRIs) display the following rank order of in vitro potency against the CYP450 isoenzyme CYP2D6 as measured by their inhibition of sparteine and/or dextromethorphan metabolism: paroxetine > fluoxetine = norfluoxetine ≥ sertraline ≥ fluvoxamine > venlafaxine. in vitro studies have shown that fluoxetine and paroxetine are more potent inhibitors of cytochrome CYP2D6 than sertraline. An approximately 10-fold increase in the Cmax and AUC24 of paroxetine and an approximately 2-fold increase in these parameters for sertraline occurred simultaneously with the desipramine concentration changes. Thus, when co-administered with 50 mg/day desipramine, sertraline had significantly less pharmacokinetic interaction than paroxetine with desipramine at the recommended starting dosages of 50 mg/day and 20 mg/day, respectively, as initially reported by Alderman et al., [81].
Second-generation antidepressants differ in their potential for pharmacokinetic drug interactions. Fluoxetine and paroxetine are potent inhibitors of CYP2D6, fluvoxamine markedly inhibits CYP1A2 and CYP2C19, and nefazodone is a substantial inhibitor of CYP3A4. Therefore, clinically relevant interactions may be expected when these antidepressants are co-administered with substrates of the pertinent isozymes, particularly those with a narrow therapeutic index. Duloxetine and bupropion are moderate inhibitors of CYP2D6, and sertraline may cause significant inhibition of this isoform, but only at high doses. Citalopram, escitalopram, venlafaxine, mirtazapine, and reboxetine are weak or negligible inhibitors of CYP isozymes in vitro and are less likely than other second-generation antidepressants to interact with co-administered medications. Second-generation antidepressants are not equivalent in their potential for pharmacokinetic drug interactions. Although interactions may be predictable in specific circumstances, use of an antidepressant with a more favorable drug-interaction profile may be justified.
The noradrenaline-selective antidepressant reboxetine in vitro is a weak inhibitor of both CYP2D6 and CYP3A4. Kuhn et al., [82] studied the pharmacokinetics of reboxetine in relation to pharmacogenetics and the effects of reboxetine compared to paroxetine treatment on the CYP2D6 and CYP3A4 phenotype. Reboxetine serum concentrations showed no correlation with the CYP2D6 genotype and the CYP2D6 phenotype, whereas paroxetine concentrations showed some dependence on CYP2D6. In contrast to in vitro investigations, indicating a major role of CYP3A4 in reboxetine metabolism, reboxetine concentrations in serum showed no correlation with the respective Dextromethorphan (DM) metabolic ratios. There was also no correlation between paroxetine concentrations and the CYP3A4 phenotype data. The CYP2C19 genotype (only heterozygosity) had no influence on reboxetine and paroxetine pharmacokinetics. There were only minor changes in the DM metabolite pattern on treatment with reboxetine and no evidence of enzyme inhibition was obtained. In contrast and as expected, paroxetine strongly inhibited CYP2D6. Thus, reboxetine treatment has no effect on the CYP2D6 genotype and no clinically relevant drug interactions involving CYP2D6 are anticipated.
Patients taking antidepressants are more likely to also be taking multiple medications, increasing the risk of adverse Drug-Drug Interactions (DDIs). Because of substantial inhibition of one or more CYP450 enzymes at therapeutic doses, the selective serotonin reuptake inhibitors fluoxetine, fluvoxamine, and paroxetine have a higher risk of CYP-mediated DDIs than citalopram, escitalopram, and sertraline, which do not substantially inhibit any CYP enzyme. In 2779 patients, 55 pairs of drugs with the potential to cause CYP-mediated DDIs occurred in 300 patients (11%), but only 26 of the patients and 6 of the drug pairs were identified by the VA Drug Alert System. Of the 461 patients receiving fluoxetine and/or paroxetine, 39 (8%) were also receiving a CYP2D6-model substrate/drug with a narrow therapeutic index, 14 (36%) of whom were receiving high enough doses to be at moderate to high risk of a serious DDI. VA patients on fluoxetine, paroxetine, and sertraline were equally likely to be on drugs whose metabolism is dependent on CYP2D6, including drugs with narrow therapeutic indices. No differences were found by Preskorn et al., [83] in doses of tricyclic antidepressants (i.e., “victim” drugs), which have narrow therapeutic indices and serious dose-dependent toxicity, when co-prescribed with fluoxetine or paroxetine vs sertraline (i.e., “perpetrator” drugs), despite predictable differences in CYP2D6-mediated clearance of these drugs.
The Selective Serotonin Reuptake Inhibitors (SSRIs) paroxetine, sertraline, and fluoxetine have varying degrees of potency in inhibiting the hepatic CYP2D6 enzyme. In an open label, parallel group study of 45 healthy volunteers, the time course of CYP2D6 inhibition of the above SSRIs was evaluated by Liston et al., [84]. Following SSRI discontinuation, calculation of a CYP2D6 inhibition half-life (t½inh) revealed the time course of fluoxetine inhibition (t½inh=7.0±1.5 days) to be significantly longer than either paroxetine (t½inh=2.9±1.9) or sertraline (t½inh=3.0±3.0), but the latter were not significantly different from each other. Time for the extrapolated DMR vs time log-linear plots to return to baseline was significantly different between fluoxetine (63.2±5.6 days) and both paroxetine (20.3±6.4 days) and sertraline (25.0±11.0 days), making the rank order (from longest to shortest) of time for CYP2D6 inhibition to dissipate: fluoxetine > sertraline ≥ paroxetine. Differences between mean baseline DMR values and measured values obtained after drug discontinuation for each drug group became non-significant on discontinuation day 5 for both paroxetine and sertraline and on discontinuation day 42 for fluoxetine. These data define the time course of a persistent effect that fluoxetine, sertraline, and paroxetine have on CYP2D6 following drug discontinuation and should be considered when initiating therapy with a CYP2D6 substrate.
Second-generation antidepressants differ in their potential for pharmacokinetic drug interactions. Fluoxetine and paroxetine are potent inhibitors of CYP2D6, fluvoxamine markedly inhibits CYP1A2 and CYP2C19, and nefazodone is a substantial inhibitor of CYP3A4. Therefore, clinically relevant interactions may be expected when these antidepressants are co-administered with substrates of the pertinent isozymes, particularly those with a narrow therapeutic index. Duloxetine and bupropion are moderate inhibitors of CYP2D6, and sertraline may cause significant inhibition of this isoform, but only at high doses. Citalopram, escitalopram, venlafaxine, mirtazapine, and reboxetine are weak or negligible inhibitors of CYP isozymes in vitro and are less likely than other second-generation antidepressants to interact with co-administered medications. Second-generation antidepressants are not equivalent in their potential for pharmacokinetic drug interactions. Although interactions may be predictable in specific circumstances, use of an antidepressant with a more favorable drug-interaction profile may be justified.
The noradrenaline-selective antidepressant reboxetine in vitro is a weak inhibitor of both CYP2D6 and CYP3A4. Kuhn et al., [82] studied the pharmacokinetics of reboxetine in relation to pharmacogenetics and the effects of reboxetine compared to paroxetine treatment on the CYP2D6 and CYP3A4 phenotype. Reboxetine serum concentrations showed no correlation with the CYP2D6 genotype and the CYP2D6 phenotype, whereas paroxetine concentrations showed some dependence on CYP2D6. In contrast to in vitro investigations, indicating a major role of CYP3A4 in reboxetine metabolism, reboxetine concentrations in serum showed no correlation with the respective Dextromethorphan (DM) metabolic ratios. There was also no correlation between paroxetine concentrations and the CYP3A4 phenotype data. The CYP2C19 genotype (only heterozygosity) had no influence on reboxetine and paroxetine pharmacokinetics. There were only minor changes in the DM metabolite pattern on treatment with reboxetine and no evidence of enzyme inhibition was obtained. In contrast and as expected, paroxetine strongly inhibited CYP2D6. Thus, reboxetine treatment has no effect on the CYP2D6 genotype and no clinically relevant drug interactions involving CYP2D6 are anticipated.
Patients taking antidepressants are more likely to also be taking multiple medications, increasing the risk of adverse Drug-Drug Interactions (DDIs). Because of substantial inhibition of one or more CYP450 enzymes at therapeutic doses, the selective serotonin reuptake inhibitors fluoxetine, fluvoxamine, and paroxetine have a higher risk of CYP-mediated DDIs than citalopram, escitalopram, and sertraline, which do not substantially inhibit any CYP enzyme. In 2779 patients, 55 pairs of drugs with the potential to cause CYP-mediated DDIs occurred in 300 patients (11%), but only 26 of the patients and 6 of the drug pairs were identified by the VA Drug Alert System. Of the 461 patients receiving fluoxetine and/or paroxetine, 39 (8%) were also receiving a CYP2D6-model substrate/drug with a narrow therapeutic index, 14 (36%) of whom were receiving high enough doses to be at moderate to high risk of a serious DDI. VA patients on fluoxetine, paroxetine, and sertraline were equally likely to be on drugs whose metabolism is dependent on CYP2D6, including drugs with narrow therapeutic indices. No differences were found by Preskorn et al., [83] in doses of tricyclic antidepressants (i.e., “victim” drugs), which have narrow therapeutic indices and serious dose-dependent toxicity, when co-prescribed with fluoxetine or paroxetine vs sertraline (i.e., “perpetrator” drugs), despite predictable differences in CYP2D6-mediated clearance of these drugs.
The Selective Serotonin Reuptake Inhibitors (SSRIs) paroxetine, sertraline, and fluoxetine have varying degrees of potency in inhibiting the hepatic CYP2D6 enzyme. In an open label, parallel group study of 45 healthy volunteers, the time course of CYP2D6 inhibition of the above SSRIs was evaluated by Liston et al., [84]. Following SSRI discontinuation, calculation of a CYP2D6 inhibition half-life (t½inh) revealed the time course of fluoxetine inhibition (t½inh=7.0±1.5 days) to be significantly longer than either paroxetine (t½inh=2.9±1.9) or sertraline (t½inh=3.0±3.0), but the latter were not significantly different from each other. Time for the extrapolated DMR vs time log-linear plots to return to baseline was significantly different between fluoxetine (63.2±5.6 days) and both paroxetine (20.3±6.4 days) and sertraline (25.0±11.0 days), making the rank order (from longest to shortest) of time for CYP2D6 inhibition to dissipate: fluoxetine > sertraline ≥ paroxetine. Differences between mean baseline DMR values and measured values obtained after drug discontinuation for each drug group became non-significant on discontinuation day 5 for both paroxetine and sertraline and on discontinuation day 42 for fluoxetine. These data define the time course of a persistent effect that fluoxetine, sertraline, and paroxetine have on CYP2D6 following drug discontinuation and should be considered when initiating therapy with a CYP2D6 substrate.
DEGREE OF CYP450 ENZYME INHIBITION OF ANTIDEPRESSANT DRUGS AT THEIR USUAL THERAPEUTIC DOSE
Amitriptyline moderately inhibits CYP2D6 and CYP2C9, and strongly inhibits CYP1A2 and CYP2C19; Citalopram is a moderate inhibitor of CYP2D6; Clomipramine is a mild inhibitor of CYP2D6 and CYP2C9, a moderate inhibitor of CYP1A2, and a strong inhibitor of CYP2C19; Desipramine moderately inhibits CYP2D6 and CYP2C19; Dothiepin is a mild inhibitor of CYP1A2 and CYP2C9, a moderate inhibitor of CYP2D6, and a strong inhibitor of CYP2C19; Doxepin is a mild inhibitor of CYP1A2 and CYP2C9, a moderate inhibitor of CYP2D6, and a strong inhibitor of CYP2C19; Duloxetine is a moderate inhibitor of CYP2D6; Escitalopram is a moderate inhibitor of CYP2D6; Fluoxetine is a strong inhibitor of CYP2D6 and CYP2C19, a moderate inhibitor of CYP2C9, and a mild inhibitor of CYP3A4; Fluvoxamine is a strong inhibitor of CYP1A2, CYP2C9 and CYP2C19, a moderate inhibitor of CYP3A4, and a mild inhibitor of CYP2D6; Imipramine is a powerful inhibitor of CYP2C19 and CYP1A2, and a moderate inhibitor of CYP2D6, CYP3A4 and CYP2C9; Nortriptyline moderately inhibits CYP2D6 and CYP2C19; Paroxetine is a strong inhibitor of CYP2D6; Sertraline is a mild inhibitor of CYP2D6; and Venlafaxine is a mild inhibitor of CYP2D6 [85] (Tables 2-5).
BIOTRANSFORMATION OF ANTIDEPRESSANTS
Tricyclic antidepressants are all hydroxylated by CYP2D6, but the tertiary amines, amitriptyline, clomipramine and imipramine, are also N-demethylated to the active metabolites, nortriptyline, N-desmethylclomipramine and desipramine, by several CYPs, including the polymorphic CYP2C19, CYP1A2 and CYP3A4. The five selective serotonin reuptake inhibitors, citalopram, fluoxetine, fluvoxamine, paroxetine and sertraline are also oxidized by the CYP enzyme system. Thus, fluoxetine, fluvoxamine and paroxetine are partially metabolized by CYP2D6, citalopram by CYP2C19 and sertraline by at least five different CYPs. Paroxetine and fluoxetine are very potent inhibitors of CYP2D6 while citalopram, fluvoxamine and sertraline are moderate inhibitors of this enzyme. Fluvoxamine is a potent inhibitor of CYP1A2 and CYP2C19 and a moderate inhibitor of CYP2C9. CYP2C19 activity is associated with the N-demethylation of tertiary amine TCA. P450 isoenzymes CYP1A2 and CYP3A4 also participate in tricyclic demethylation. The tertiary amine imipramine is typically metabolized by demethylation to the secondary active metabolite desipramine. Desipramine is also a tricyclic drug by itself. Both tertiary and secondary amines are metabolized, mainly through hydroxylation by CYP2D6, to 2-OH-imipramine and 2-OH-desipramine. Hydroxy-metabolites of TCAs have similar or shorter half-lives and smaller distribution volumes than their parent compounds. Glucuronide conjugation of TCA hydroxylated metabolites improves their water solubility and therefore promotes more efficient renal excretion. The dose range of many tricyclic antidepressants is between 25-300 mg/day. Genetically-based differences in the activity of CYP2C19 and CYP2D6 account in large part for the wide interindividual variability in the drug’s metabolism and plasma concentration. Depending on the antidepressant, CYP2C19 poor metabolizers, as well as CYP2D6 poor metabolizers, may generate plasma drug levels several fold higher during routine dosing. Toxic effects, such as agitation, confusion, and pacing, are seen in patients with TCA plasma concentrations greater than 450-500 ng/mL. Major toxicity and death is associated with concentrations above 1000 ng/mL. In terms of drug interactions, desipramine is not a strong inhibitor of CYP2D6, but imipramine significantly inhibits CYP2C19 and CYP1A2 [1,86-88].
Genetic variation in drug-metabolizing enzymes such as CYP2D6 has been postulated to underlie antidepressant intolerance (pharmacokinetic effect). However, variation in genes encoding serotonin receptors could also explain antidepressant side-effects (pharmacodynamic effect). Survival analysis showed that discontinuations due to paroxetine-induced side-effects were strongly associated with the HTR2A C/C genotype. There was a significant linear relationship between the number of C alleles and the probability of discontinuation. Side-effect severity in paroxetine-treated patients with the C/C genotype was also greater. In contrast, HTR2A 102 T/C genotype had no effect on mirtazapine side-effects. CYP2D6 genotype did not predict treatment outcome for either medication. Pharmacodynamic differences among patients due to variant 5-HT2A receptors appear to be more important than pharmacokinetic variation in determining paroxetine intolerance. Pharmacogenetic markers may be useful in predicting antidepressant treatment outcome [1,89,90].
Genetic variation in drug-metabolizing enzymes such as CYP2D6 has been postulated to underlie antidepressant intolerance (pharmacokinetic effect). However, variation in genes encoding serotonin receptors could also explain antidepressant side-effects (pharmacodynamic effect). Survival analysis showed that discontinuations due to paroxetine-induced side-effects were strongly associated with the HTR2A C/C genotype. There was a significant linear relationship between the number of C alleles and the probability of discontinuation. Side-effect severity in paroxetine-treated patients with the C/C genotype was also greater. In contrast, HTR2A 102 T/C genotype had no effect on mirtazapine side-effects. CYP2D6 genotype did not predict treatment outcome for either medication. Pharmacodynamic differences among patients due to variant 5-HT2A receptors appear to be more important than pharmacokinetic variation in determining paroxetine intolerance. Pharmacogenetic markers may be useful in predicting antidepressant treatment outcome [1,89,90].
PLASMA CONCENTRATIONS
Studies focused on Polymorphisms of the Cytochrome-P450 (CYP) isoenzymes 2C9, 2C19 and 2D6 and their association with plasma concentrations within a typical clinical setting indicate that CYP2D6 poor metabolizer genotype and co-medication with inhibitors of CYP2D6 were associated with higher plasma concentrations than the drug-specific median plasma concentration when normalized to dose; plasma concentrations of CYP2C19 extensive metabolizers and smokers were significantly lower than the drug-specific median. CYP2D6 poor metabolizers experienced side-effects. Response was not associated with plasma concentrations above or below the lower limit of a presumed therapeutic range. These data indicate a significant influence of the CYP2D6 genotype, minor influence of the CYP2C19 genotype and no influence of the CYP2C9 genotype on plasma concentrations of patients taking mainly second-generation antidepressants [1,91].
Relative effects of duloxetine, escitalopram, and sertraline on the functional activity of the drug-metabolizing CYP2D6 enzyme, as assessed by changes in the pharmacokinetics of the CYP2D6 model substrate drug metoprolol, revealed the following rank order for the change in metoprolol area under the plasma concentration-time curve: duloxetine (180%) > escitalopram (89%) > sertraline (48% and 67%). Compared with sertraline, duloxetine produced statistically significantly larger changes in metoprolol pharmacokinetic parameters. The changes produced by escitalopram and sertraline were not statistically different [83].
in vivo, inhibitory potency is affected by the plasma concentration of the free (unbound) drug, a potentially important consideration since many CYP2D6-metabolized drugs exhibit nonlinear (saturable) kinetics, and by the presence of metabolites, which might accumulate and interact with the CYP system. Under steady-state conditions, paroxetine and fluoxetine are approximately clinically equipotent inhibitors of CYP2D6 in vivo (as determined through their effects on desipramine metabolism); sertraline, in contrast, shows lower steady-state plasma concentrations than fluoxetine and, hence, a less pronounced inhibition of CYP2D6. Of the drugs metabolized by CYP2D6, secondary amine tricyclic antidepressants, antipsychotics (eg: phenothiazines, and risperidone), codeine, some antiarrhythmics (eg: flecainide) and β-blockers form the focus of clinical attention with regard to their potential interactions with the SSRIs. Co-administration of desipramine and fluoxetine (20 mg/day) at steady-state produced an approximately 4-fold elevation in peak plasma desipramine concentrations, while the long half-life of the active metabolite norfluoxetine was responsible for a significant and long-lasting (approximately 3 weeks) elevation of plasma desipramine concentrations after discontinuation of fluoxetine. Similarly, co-administration of desipramine with paroxetine produced an approximately 3-fold increase in plasma desipramine concentration. In contrast, co-administration of desipramine and sertraline (50 mg/day) for 4 weeks resulted in a considerably more modest (approximately 30%) elevation in plasma desipramine concentrations. Co-administration of fluoxetine (60 mg/day, as a loading dose) [equivalent to serum concentrations obtained with 20 mg/day at steady-state] with imipramine or desipramine resulted in approximately 3- to 4-fold increases in plasma Area Under the Curve (AUC) values for both imipramine and desipramine (illustrating a significant drug interaction potential at multiple isoenzymes). Consistent with its minimal in vitro effect on CYP2D6, fluvoxamine shows minimal in vivo pharmacokinetic interaction with desipramine, but does interact with imipramine (approximately 3- to 4-fold increase in AUC) through inhibition of CYP3A3/4, CYP1A2, and CYP2C19 [80,84].
Relative effects of duloxetine, escitalopram, and sertraline on the functional activity of the drug-metabolizing CYP2D6 enzyme, as assessed by changes in the pharmacokinetics of the CYP2D6 model substrate drug metoprolol, revealed the following rank order for the change in metoprolol area under the plasma concentration-time curve: duloxetine (180%) > escitalopram (89%) > sertraline (48% and 67%). Compared with sertraline, duloxetine produced statistically significantly larger changes in metoprolol pharmacokinetic parameters. The changes produced by escitalopram and sertraline were not statistically different [83].
in vivo, inhibitory potency is affected by the plasma concentration of the free (unbound) drug, a potentially important consideration since many CYP2D6-metabolized drugs exhibit nonlinear (saturable) kinetics, and by the presence of metabolites, which might accumulate and interact with the CYP system. Under steady-state conditions, paroxetine and fluoxetine are approximately clinically equipotent inhibitors of CYP2D6 in vivo (as determined through their effects on desipramine metabolism); sertraline, in contrast, shows lower steady-state plasma concentrations than fluoxetine and, hence, a less pronounced inhibition of CYP2D6. Of the drugs metabolized by CYP2D6, secondary amine tricyclic antidepressants, antipsychotics (eg: phenothiazines, and risperidone), codeine, some antiarrhythmics (eg: flecainide) and β-blockers form the focus of clinical attention with regard to their potential interactions with the SSRIs. Co-administration of desipramine and fluoxetine (20 mg/day) at steady-state produced an approximately 4-fold elevation in peak plasma desipramine concentrations, while the long half-life of the active metabolite norfluoxetine was responsible for a significant and long-lasting (approximately 3 weeks) elevation of plasma desipramine concentrations after discontinuation of fluoxetine. Similarly, co-administration of desipramine with paroxetine produced an approximately 3-fold increase in plasma desipramine concentration. In contrast, co-administration of desipramine and sertraline (50 mg/day) for 4 weeks resulted in a considerably more modest (approximately 30%) elevation in plasma desipramine concentrations. Co-administration of fluoxetine (60 mg/day, as a loading dose) [equivalent to serum concentrations obtained with 20 mg/day at steady-state] with imipramine or desipramine resulted in approximately 3- to 4-fold increases in plasma Area Under the Curve (AUC) values for both imipramine and desipramine (illustrating a significant drug interaction potential at multiple isoenzymes). Consistent with its minimal in vitro effect on CYP2D6, fluvoxamine shows minimal in vivo pharmacokinetic interaction with desipramine, but does interact with imipramine (approximately 3- to 4-fold increase in AUC) through inhibition of CYP3A3/4, CYP1A2, and CYP2C19 [80,84].
OTHER CONSIDERATIONS
Brøsen [92] reviewed the pharmacological properties and differences of SSRIs. SSRIs differ from each other with regard to their chemical structure, their pharmacokinetics and their potential for causing pharmacokinetic interactions through inhibition of species of the CYP enzyme system. Fluvoxamine, but not citalopram, fluoxetine, paroxetine or sertraline, is a potent inhibitor of CYP1A2. Fluvoxamine has interactions with other drugs eliminated by CYP1A2, including caffeine, clozapine, olanzapine, theophylline, propranolol and tacrine. CYP2C19 is the source of the S-mephenytoin oxidation polymorphism. About 2% of whites are poor metabolizers in whom CYP2C19 is not expressed. Poor metabolizers have an impaired elimination of citalopram, among other drugs. Although not metabolized by CYP2C19, fluvoxamine is still a potent inhibitor of the enzyme. The same applies to fluoxetine. CYP2D6 only makes up about 2-5% of the total P450 in the human liver, but is the major enzyme catalyzing more than 30 clinically-used drugs, including all of the tricyclic antidepressants, several neuroleptics, opiates, β-blockers, antiarrhythmics and among the SSRIs N-desmethylcitalopram, fluvoxamine, fluoxetine and paroxetine but not sertraline. All of the SSRIs inhibit CYP2D6 but fluoxetine, norfluoxetine and paroxetine are particularly potent inhibitors. CYP3A4 is the most abundant human CYP450, but most of the SSRIs, with the exception of norfluoxetine, do not inhibit this enzyme, and interactions with SSRIs and CYP3A4 appear not to be significant.
Baumann [93] reviewed the pharmacological features of SSRIs in the elderly. Citalopram, fluoxetine, fluvoxamine, paroxetine and sertraline are the selective serotonin reuptake inhibiting antidepressants available. They differ by their chemical structure, metabolism and pharmacokinetics. The pharmacokinetics of drugs may be modified in elderly patients at different levels: absorption, distribution, metabolism and excretion. In these patients, depending on the SSRI used, it is recommended to adapt the dose of the antidepressant. Lower doses should be used for citalopram, paroxetine and probably also for sertraline, when therapy is initiated. No clear evidence was found for fluoxetine and fluvoxamine concerning age-dependent metabolism. As elderly depressive patients may also suffer from somatic diseases, this should be considered in the choice of the dose of an SSRI, as for some of them, elimination is decreased in hepatic (citalopram, fluoxetine, fluvoxamine, sertraline) or renal (paroxetine) impairments.
An antidepressant for use in patients receiving concomitant drug treatment, over-the-counter medications, or herbal products should lack CYP3A4 inductive or inhibitory activity, in order to provide the least likelihood of a drug-drug interaction. DeVane et al., [94] studied the potential of 4 diverse antidepressants (venlafaxine, nefazodone, sertraline, and fluoxetine) to inhibit or induce CYP3A4. Compared to baseline, venlafaxine, sertraline, and fluoxetine caused no apparent inhibition or induction of erythromycin metabolism. For nefazodone, a statistically significant inhibition was observed. Nefazodone was also the only antidepressant that caused a significant change in alprazolam disposition, decreasing its area under the concentration-vs-time curve, and increasing its elimination half-life (16.4 vs 12.3 hours) compared with values at baseline. No significant differences were found in the pharmacokinetics of alprazolam with any of the other antidepressants tested. These results demonstrate in vivo that, unlike nefazodone, venlafaxine, sertraline, and fluoxetine do not possess significant metabolic inductive or inhibitory effects on CYP3A4.
Haduch et al., [95] investigated the influence of Tricyclic Antidepressants (TCAs: imipramine, amitriptyline, clomipramine, and desipramine), Selective Serotonin Reuptake Inhibitors (SSRIs: fluoxetine and sertraline) and novel antidepressant drugs (mirtazapine and nefazodone) on the activity of CYP3A measured as a rate of testosterone 2β- and 6β-hydroxylation. These antidepressants, added to control liver microsomes, produced some inhibitory effects on CYP3A activity, which were very weak, modest (clomipramine and sertraline) or moderate (nefazodone and fluoxetine). Mirtazapine did not display this kind of properties. One-day exposure of rats to TCAs substantially decreased the activity of CYP3A in liver microsomes, which was maintained during chronic treatment. The observed decreases in the enzyme activity were in contrast to the increased CYP3A protein level found after chronic treatment with TCAs. Sertraline increased the activity of the enzyme after its prolonged administration, and its effect correlated positively with the observed elevation in CYP3A protein level. Fluoxetine, mirtazapine and nefazodone did not change the activity of CYP3A in liver microsomes after their administration to rats. Three different mechanisms of the antidepressant-CYP3A interaction were postulated: (i) a direct inhibition of CYP3A by nefazodone, SSRIs and clomipramine, shown in vitro, with the inhibitory effect of nefazodone being the strongest, but weaker than the effects of this drug on human CYP3A4; (ii) in vivo inhibition of CYP3A produced by 1 day and maintained during chronic treatment with TCAs, which suggests inactivation of the enzyme by reactive metabolites; (iii) in vivo induction by sertraline of CYP3A produced only by chronic treatment with the antidepressant, which suggests its influence on the enzyme regulation.
Pharmacological treatment of depression in old age is associated with an increased risk of adverse pharmacokinetic and pharmacodynamic drug interactions. Elderly patients may have multiple disease states and, therefore, may require a variety of other drugs. In addition to polypharmacy, other factors such as age-related physiological changes, diseases, genetic constitution and diet may alter drug response and therefore predispose elderly patients to adverse effects and drug interactions. Antidepressant drugs currently available differ in their potential for drug interactions. In general, older compounds, such as TCAs and MAOIs, have a higher potential for interactions than newer compounds, such as SSRIs and other relatively novel agents with a more specific mechanism of action. In particular, TCAs and MAOIs are associated with clinically significant pharmacodynamic interactions with many medications frequently prescribed to elderly patients. TCAs may be susceptible to pharmacokinetic interactions when given in combination with inhibitors or inducers of the CYP isoenzymes involved in their metabolism. Because of a more selective mechanism of action, newer antidepressants have a low potential for pharmacodynamic drug interactions. However, the possibility of the serotonin syndrome should be taken into account when drugs affecting serotonergic transmission, such as SSRIs, venlafaxine or nefazodone, are co-administered with other serotonergic agents. Newer agents have a differential potential for pharmacokinetic interactions because of their selective effects on CYP isoenzymes. Within the group of SSRIs, fluoxetine and paroxetine are potent inhibitors of CYP2D6, while fluvoxamine predominantly affects CYP1A2 and CYP2C19 activity. Therefore, these agents should be closely monitored or avoided in elderly patients treated with substrates of these isoforms, especially those with a narrow therapeutic index. Citalopram and sertraline have a low inhibitory activity on different drug-metabolizing enzymes and appear particularly suitable in an elderly population. Among other newer antidepressants, nefazodone is a potent inhibitor of CYP3A4, and its combination with substrates of this isoform should be avoided, according to Spina and Scordo [96].
Baumann [93] reviewed the pharmacological features of SSRIs in the elderly. Citalopram, fluoxetine, fluvoxamine, paroxetine and sertraline are the selective serotonin reuptake inhibiting antidepressants available. They differ by their chemical structure, metabolism and pharmacokinetics. The pharmacokinetics of drugs may be modified in elderly patients at different levels: absorption, distribution, metabolism and excretion. In these patients, depending on the SSRI used, it is recommended to adapt the dose of the antidepressant. Lower doses should be used for citalopram, paroxetine and probably also for sertraline, when therapy is initiated. No clear evidence was found for fluoxetine and fluvoxamine concerning age-dependent metabolism. As elderly depressive patients may also suffer from somatic diseases, this should be considered in the choice of the dose of an SSRI, as for some of them, elimination is decreased in hepatic (citalopram, fluoxetine, fluvoxamine, sertraline) or renal (paroxetine) impairments.
An antidepressant for use in patients receiving concomitant drug treatment, over-the-counter medications, or herbal products should lack CYP3A4 inductive or inhibitory activity, in order to provide the least likelihood of a drug-drug interaction. DeVane et al., [94] studied the potential of 4 diverse antidepressants (venlafaxine, nefazodone, sertraline, and fluoxetine) to inhibit or induce CYP3A4. Compared to baseline, venlafaxine, sertraline, and fluoxetine caused no apparent inhibition or induction of erythromycin metabolism. For nefazodone, a statistically significant inhibition was observed. Nefazodone was also the only antidepressant that caused a significant change in alprazolam disposition, decreasing its area under the concentration-vs-time curve, and increasing its elimination half-life (16.4 vs 12.3 hours) compared with values at baseline. No significant differences were found in the pharmacokinetics of alprazolam with any of the other antidepressants tested. These results demonstrate in vivo that, unlike nefazodone, venlafaxine, sertraline, and fluoxetine do not possess significant metabolic inductive or inhibitory effects on CYP3A4.
Haduch et al., [95] investigated the influence of Tricyclic Antidepressants (TCAs: imipramine, amitriptyline, clomipramine, and desipramine), Selective Serotonin Reuptake Inhibitors (SSRIs: fluoxetine and sertraline) and novel antidepressant drugs (mirtazapine and nefazodone) on the activity of CYP3A measured as a rate of testosterone 2β- and 6β-hydroxylation. These antidepressants, added to control liver microsomes, produced some inhibitory effects on CYP3A activity, which were very weak, modest (clomipramine and sertraline) or moderate (nefazodone and fluoxetine). Mirtazapine did not display this kind of properties. One-day exposure of rats to TCAs substantially decreased the activity of CYP3A in liver microsomes, which was maintained during chronic treatment. The observed decreases in the enzyme activity were in contrast to the increased CYP3A protein level found after chronic treatment with TCAs. Sertraline increased the activity of the enzyme after its prolonged administration, and its effect correlated positively with the observed elevation in CYP3A protein level. Fluoxetine, mirtazapine and nefazodone did not change the activity of CYP3A in liver microsomes after their administration to rats. Three different mechanisms of the antidepressant-CYP3A interaction were postulated: (i) a direct inhibition of CYP3A by nefazodone, SSRIs and clomipramine, shown in vitro, with the inhibitory effect of nefazodone being the strongest, but weaker than the effects of this drug on human CYP3A4; (ii) in vivo inhibition of CYP3A produced by 1 day and maintained during chronic treatment with TCAs, which suggests inactivation of the enzyme by reactive metabolites; (iii) in vivo induction by sertraline of CYP3A produced only by chronic treatment with the antidepressant, which suggests its influence on the enzyme regulation.
Pharmacological treatment of depression in old age is associated with an increased risk of adverse pharmacokinetic and pharmacodynamic drug interactions. Elderly patients may have multiple disease states and, therefore, may require a variety of other drugs. In addition to polypharmacy, other factors such as age-related physiological changes, diseases, genetic constitution and diet may alter drug response and therefore predispose elderly patients to adverse effects and drug interactions. Antidepressant drugs currently available differ in their potential for drug interactions. In general, older compounds, such as TCAs and MAOIs, have a higher potential for interactions than newer compounds, such as SSRIs and other relatively novel agents with a more specific mechanism of action. In particular, TCAs and MAOIs are associated with clinically significant pharmacodynamic interactions with many medications frequently prescribed to elderly patients. TCAs may be susceptible to pharmacokinetic interactions when given in combination with inhibitors or inducers of the CYP isoenzymes involved in their metabolism. Because of a more selective mechanism of action, newer antidepressants have a low potential for pharmacodynamic drug interactions. However, the possibility of the serotonin syndrome should be taken into account when drugs affecting serotonergic transmission, such as SSRIs, venlafaxine or nefazodone, are co-administered with other serotonergic agents. Newer agents have a differential potential for pharmacokinetic interactions because of their selective effects on CYP isoenzymes. Within the group of SSRIs, fluoxetine and paroxetine are potent inhibitors of CYP2D6, while fluvoxamine predominantly affects CYP1A2 and CYP2C19 activity. Therefore, these agents should be closely monitored or avoided in elderly patients treated with substrates of these isoforms, especially those with a narrow therapeutic index. Citalopram and sertraline have a low inhibitory activity on different drug-metabolizing enzymes and appear particularly suitable in an elderly population. Among other newer antidepressants, nefazodone is a potent inhibitor of CYP3A4, and its combination with substrates of this isoform should be avoided, according to Spina and Scordo [96].
CONCLUSION
The optimization of CNS therapeutics requires the establishment of new postulates regarding (i) the costs of medicines, (ii) the assessment of protocols for multifactorial treatment in chronic disorders, (iii) the implementation of novel therapeutics addressing causative factors, and (iv) the setting-up of pharmacogenomic strategies for drug development. Pharmacogenomics accounts for 30-90% variability in pharmacokinetics and pharmacodynamics. Personalized therapeutics based on individual genomic profiles implies the characterization of 5 types of gene clusters: (i) genes associated with disease pathogenesis; (ii) genes associated with the mechanism of action of drugs; (iii) genes associated with drug metabolism (phase I and II reactions); (iv) genes associated with drug transporters; and (v) pleiotropic genes involved in multifaceted cascades and metabolic reactions.
Priority areas for pharmacogenetic research are the prediction of serious adverse reactions and the establishment of variation in efficacy. Both requirements are necessary in depression, to cope with efficacy and safety issues associated with current antidepressant drugs, and new CNS drugs as well.
With regard to the future of pharmacogenomics as a practical discipline to efficiently optimize therapeutics, several issues should be addressed: (i) the education of physicians in medical genomics and pharmacogenomics is fundamental (less than 2% of the members of the medical community are familiar with genomic science); (ii) genomic screening of gene clusters involved in pharmacogenomic outcomes must become a clinical routine (whithout genetic testing there is no pharmacogenetics); (iii) each patient must be a carrier of a pharmacogenetic card indicating what kind of drugs he/she can take and which medications he/she should avoid; (iv) Regulatory Agencies should request pharmacogenetic data from the pharmaceutical industry when applying for drug approval; (v) pharmacogenetic data must be incorporated into the patient information leaflet and the pharmaceutical vade mecum; and (vi) new guidelines for daily praxis, such as that of the first World Guide for Drug Use and Pharmacogenomics, will facilitate the understanding of the relationship between drugs and genes (and vice versa) to make drug prescription a real personalized procedure.
Priority areas for pharmacogenetic research are the prediction of serious adverse reactions and the establishment of variation in efficacy. Both requirements are necessary in depression, to cope with efficacy and safety issues associated with current antidepressant drugs, and new CNS drugs as well.
With regard to the future of pharmacogenomics as a practical discipline to efficiently optimize therapeutics, several issues should be addressed: (i) the education of physicians in medical genomics and pharmacogenomics is fundamental (less than 2% of the members of the medical community are familiar with genomic science); (ii) genomic screening of gene clusters involved in pharmacogenomic outcomes must become a clinical routine (whithout genetic testing there is no pharmacogenetics); (iii) each patient must be a carrier of a pharmacogenetic card indicating what kind of drugs he/she can take and which medications he/she should avoid; (iv) Regulatory Agencies should request pharmacogenetic data from the pharmaceutical industry when applying for drug approval; (v) pharmacogenetic data must be incorporated into the patient information leaflet and the pharmaceutical vade mecum; and (vi) new guidelines for daily praxis, such as that of the first World Guide for Drug Use and Pharmacogenomics, will facilitate the understanding of the relationship between drugs and genes (and vice versa) to make drug prescription a real personalized procedure.
REFERENCES
- Cacabelos R (2012) World Guide for Drug Use and Pharmacogenomics. EuroEspes Publishing, Corunna, Spain.
- Meyer UA (2004) Pharmacogenetics - five decades of therapeutic lessons from genetic diversity. Nat Rev Genet 5: 669-676.
- Kalow W, Meyer UA, Tyndale RF (2001) Pharmacogenomics. Marcel Dekker, New York, USA.
- Cacabelos R (2012) Pharmacogenomics of Central Nervous System (CNS) drugs. Drug Dev Res 73: 461-476.
- Cacabelos R, Cacabelos P, Aliev G (2013) Genomics of schizophrenia and pharmacogenomics of antipsychotic drugs. Open J Psychiatry 3: 46-139.
- Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, et al. (2001) Initial sequencing and analysis of the human genome. Nature 409: 860-921.
- Pennisi E (2001) The human genome. Science 291: 1177-1180.
- Various authors (2006) Human Genome. Nature 1: 7-305.
- Altman RB, Flockhart D, Goldstein DB (2012) Principles of Pharmacogenetics and Pharmacogenomics. Cambridge University Press, New York, USA.
- American College of Clinical Pharmacy (2004) Pharmacogenomics: Applications to Patients Care. American College of Clinical Pharmacy, Kansas City, MO, USA.
- Humma LM, Cavallari LM, Ellingrod VL, Kolesar JM (2005) Pharmaco-Genomics Handbook. Lexi-Comp, Hudson, Ohio, USA.
- Cohen N (2008) Pharmacogenomics and Personalized Medicine. Humana Press, Totowa, NJ, USA.
- Hall IP, Pirmohamed M (2006) Pharmacogenetics. Informa Healthcare, Taylor & Francis, New York, USA.
- Yan Q (2008) Pharmacogenomics in drug discovery and development. Preface. Methods Mol Biol 448: v-vii.
- Helton SG, Lohoff FW (2015) Serotonin pathway polymorphisms and the treatment of major depressive disorder and anxiety disorders. Pharmacogenomics 16: 541-553.
- Cacabelos R, Cacabelos P, Torrellas C, Tellado I, Carril JC (2014) Pharmacogenomics of Alzheimer’s disease: novel therapeutic strategies for drug development. Methods Mol Biol 1175: 323-556.
- Cacabelos R (2012) The metabolomic paradigm of pharmacogenomics in complex disorders. Metabolomics 2: 119.
- Cacabelos R, Martinez-Bouza R, Carril JC, Fernandez-Novoa L, Lombardi V, et al. (2012) Genomics and pharmacogenomics of brain disorders. Curr Pharm Biotechnol 13: 674-725.
- Fabbri C, Serretti A (2015) Pharmacogenetics of major depressive disorder: top genes and pathways toward clinical applications. Curr Psychiatry Rep 17: 50.
- Torrellas C, Carril JC, Cacabelos R (2014) Pharmacogenetics of Antidepressant drugs: Optimizing prescription. Drug Metabol Drug Interact 29: 91-92.
- Torrellas C, Risso A, Carril JC, Cacabelos R (2015) Pharmacogenetics-related optimization of antidepressant use. GenT 10.
- Cacabelos R, Takeda M (2006) Pharmacogenomics, nutrigenomics and future therapeutics in Alzheimer’s disease. Drugs Fut 31: 5.
- Lobo AL, Tomás E, da Silva FP, Barbot J, Raposo T (2001) Anemia Neonatal Grave por Hemorragia Feto-Materna Caso Clínico. Acta Pediatr Port 32: 395-397.
- Cacabelos R (2007) Molecular pathology and pharmacogenomics in Alzheimer’s disease: polygenic-related effects of multifactorial treatments on cognition, anxiety and depression. Methods Find Exp Clin Pharmacol 29: 1-91.
- Cacabelos R, Fernández-Novoa L, Martínez-Bouza R, McKay A, Carril JC, et al. (2010) Future Trends in the Pharmacogenomics of Brain Disorders and Dementia: Influence of APOE and CYP2D6 Variants. Pharmaceuticals 3: 3040-3100.
- Sullivan PF, Neale MC, Kendler KS (2000) Genetic epidemiology of major depression: review and meta-analysis. Am J Psychiatry 157: 1552-1562.
- Marazita ML, Neiswanger K, Cooper M, Zubenko GS, Giles DE, et al. (1997) Genetic segregation analysis of early-onset recurrent unipolar depression. Am J Hum Genet 61: 1370-1378.
- Binder EB, Salyakina D, Lichtner P, Wochnik GM, Ising M, et al. (2004) Polymorphisms in FKBP5 are associated with increased recurrence of depressive episodes and rapid response to antidepressant treatment. Nat Genet 36: 1319-1325.
- Zhang X, Gainetdinov RR, Beaulieu JM, Sotnikova TD, Burch LH, et al. (2005) Loss-of-function mutation in tryptophan hydroxylase-2 identified in unipolar major depression. Neuron 45: 11-16.
- Abkevich V, Camp NJ, Hensel CH, Neff CD, Russell DL, et al. (2003) Predisposition locus for major depression at chromosome 12q22-12q23.2. Am J Hum Genet 73: 1271-1281.
- McMahon FJ, Buervenich S, Charney D, Lipsky R, Rush AJ, et al. (2006) Variation in the gene encoding the serotonin 2A receptor is associated with outcome of antidepressant treatment. Am J Hum Genet 78: 804-814.
- Holmans P, Zubenko GS, Crowe RR, DePaulo JR Jr, Scheftner WA, et al. (2004) Genomewide significant linkage to recurrent, early-onset major depressive disorder on chromosome 15q. Am J Hum Genet 74: 1154-1167.
- St Clair D, Blackwood D, Muir W, Carothers A, Walker M, et al. (1990) Association within a family of a balanced autosomal translocation with major mental illness. Lancet 336: 13-16.
- Caroppo P, Le Ber I, Clot F, Rivaud-Péchoux S, Camuzat A, et al. (2014) DCTN1 mutation analysis in families with progressive supranuclear palsy-like phenotypes. JAMA Neurol 71: 208-215.
- Wider C, Dachsel JC, Farrer MJ, Dickson DW, Tsuboi Y, et al. (2010) Elucidating the genetics and pathology of Perry syndrome. J Neurol Sci 289: 149-154.
- Jansen R, Penninx BW, Madar V, Xia K, Milaneschi Y, et al. (2015) Gene expression in major depressive disorder. Mol Psychiatry.
- Genetics of Personality Consortium, de Moor MH, van den Berg SM, Verweij KJ, Krueger RF, et al. (2015) Meta-analysis of Genome-wide Association Studies for Neuroticism, and the Polygenic Association With Major Depressive Disorder. JAMA Psychiatry 72: 642-650.
- Cai N, Chang S, Li Y, Li Q, Hu J, et al. (2015) Molecular signatures of major depression. Curr Biol 25: 1146-1156.
- Czarny P, Kwiatkowski D, Galecki P, Talarowska M, Orzechowska A, et al. (2015) Association between single nucleotide polymorphisms of MUTYH, hOGG1 and NEIL1 genes, and depression. J Affect Disord 184: 90-96.
- Giniatullina A, Maroteaux G, Geerts CJ, Koopmans B, Loos M, et al. (2015) Functional characterization of the PCLO p.Ser4814Ala variant associated with major depressive disorder reveals cellular but not behavioral differences. Neuroscience 300: 518-538.
- Milaneschi Y, Lamers F, Peyrot WJ, Abdellaoui A, Willemsen G, et al. (2015) Polygenic dissection of major depression clinical heterogeneity. Mol Psychiatry.
- Bagheri A, Kamalidehghan B, Haghshenas M, Azadfar P, Akbari L, et al. (2015) Prevalence of the CYP2D6*10 (C100T), *4 (G1846A), and *14 (G1758A) alleles among Iranians of different ethnicities. Drug Des Devel Ther 9: 2627-2634.
- Marquez B, Van Bambeke F (2011) ABC multidrug transporters: target for modulation of drug pharmacokinetics and drug-drug interactions. Curr Drug Targets 12: 600-620.
- Haufroid V (2011) Genetic polymorphisms of ATP-binding cassette transporters ABCB1 and ABCC2 and their impact on drug disposition. Curr Drug Targets 12: 631-646.
- Hosoya K, Tachikawa M (2011) Roles of organic anion/cation transporters at the blood-brain and blood-cerebrospinal fluid barriers involving uremic toxins. Clin Exp Nephrol 15: 478-485.
- Carl SM, Lindley DJ, Das D, Couraud PO, Weksler BB, et al. (2010) ABC and SLC transporter expression and proton oligopeptide transporter (POT) mediated permeation across the human blood--brain barrier cell line, hCMEC/D3. Mol Pharm 7: 1057-1068.
- Cacabelos R, Fernandez-Novoa L, Lombardi V, Kubota Y, Takeda M (2005) Molecular genetics of Alzheimer’s disease and aging. Methods Find Exp Clin Pharmacol 27: 1-573.
- Takeda M, Martínez R, Kudo T, Tanaka T, Okochi M, et al. (2010) Apolipoprotein E and central nervous system disorders: reviews of clinical findings. Psychiatry Clin Neurosci 64: 592-607.
- Cacabelos R (2003) The application of functional genomics to Alzheimer’s disease. Pharmacogenomics 4: 597-621.
- Cacabelos R (2008) Pharmacogenomics in Alzheimer’s disease. Methods Mol Biol 448: 213-357.
- Luciano M, Pujals AM, Marioni RE, Campbell A, Hayward C, et al. (2015) Current versus lifetime depression, APOE variation, and their interaction on cognitive performance in younger and older adults. Psychosom Med 77: 480-492.
- Qureshi IA, Mehler MF (2011) Advances in epigenetics and epigenomics for neurodegenerative diseases. Curr Neurol Neurosci Rep 11: 464-473.
- Erickson MA, Banks WA (2013) Blood-brain barrier dysfunction as a cause and consequence of Alzheimer’s disease. J Cereb Blood Flow Metab 33: 1500-1513.
- Zhang GL, Zhang WG, Du Y, Yao L, Sun H, et al. (2013) Edaravone ameliorates oxidative damage associated with Aβ25-35 treatment in PC12 cells. J Mol Neurosci 50: 494-503.
- Shen YE, Wang Y, Yu GC, Liu C, Zhang ZY, et al. (2013) Effects of edaravone on amyloid-β precursor protein processing in SY5Y-APP695 cells. Neurotox Res 24: 139-147.
- Knowles JK, Simmons DA, Nguyen TV, Vander Griend L, Xie Y, et al. (2013) Small molecule p75NTR ligand prevents cognitive deficits and neurite degeneration in an Alzheimer's mouse model. Neurobiol Aging 34: 2052-2063.
- Wang T, Huang Y, Zhang M, Wang L, Wang Y, et al. (2013) [Gly14]-Humanin offers neuroprotection through glycogen synthase kinase-3β inhibition in a mouse model of intracerebral hemorrhage. Behav Brain Res 247: 132-139.
- Sakurai T, Kitadate K, Nishioka H, Fujii H, Ogasawara J, et al. (2013) Oligomerised lychee fruit-derived polyphenol attenuates cognitive impairment in senescence-accelerated mice and endoplasmic reticulum stress in neuronal cells. Br J Nutr 28: 1-10.
- Quitschke WW, Steinhauff N, Rooney J (2013) The effect of cyclodextrin-solubilized curcuminoids on amyloid plaques in Alzheimer transgenic mice: brain uptake and metabolism after intravenous and subcutaneous injection. Alzheimers Res Ther 5: 16.
- Geekiyanage H, Upadhye A, Chan C (2013) Inhibition of serine palmitoyltransferase reduces Aβ and tau hyperphosphorylation in a murine model: a safe therapeutic strategy for Alzheimer’s disease. Neurobiol Aging 34: 2037-2051.
- Jarmu?a A, St?pkowski D (2013) The β-sheet breakers and π-stacking. J Pept Sci 19: 345-349.
- Aso E, Juvés S, Maldonado R, Ferrer I (2013) CB2 cannabinoid receptor agonist ameliorates Alzheimer-like phenotype in AβPP/PS1 mice. J Alzheimers Dis 35: 847-858.
- Fernandes RA, Ingle AB (2013) Arundic acid a potential neuroprotective agent: biological development and syntheses. Curr Med Chem 20: 2315-2329.
- Yao ZG, Zhang L, Liang L, Liu Y, Yang YJ, et al. (2013) The effect of PN-1, a Traditional Chinese Prescription, on the Learning and Memory in a Transgenic Mouse Model of Alzheimer’s Disease. Evid Based Complement Alternat Med 2013: 518421.
- Chen N, Yang M, Guo J, Zhou M, Zhu C, et al. (2013) Cerebrolysin for vascular dementia. Cochrane Database Syst Rev 1: CD008900.
- Alvarez XA, Cacabelos R, Sampedro C, Couceiro V, Aleixandre M, et al. (2011) Combination treatment in Alzheimer’s disease: results of a randomized, controlled trial with cerebrolysin and donepezil. Curr Alzheimer Res 8: 583-591.
- Brinton RD (2013) Neurosteroids as regenerative agents in the brain: therapeutic implications. Nat Rev Endocrinol 9: 241-250.
- Abdul-Hay SO, Lane AL, Caulfield TR, Claussin C, Bertrand J, et al. (2013) Optimization of peptide hydroxamate inhibitors of insulin-degrading enzyme reveals marked substrate-selectivity. J Med Chem 56: 2246-2255.
- Devi L, Ohno M (2013) Effects of levetiracetam, an antiepileptic drug, on memory impairments associated with aging and Alzheimer’s disease in mice. Neurobiol Learn Mem 102: 7-11.
- Cotroneo AM, Castagna A, Putignano S, Lacava R, Fantò F, et al. (2013) Effectiveness and safety of citicoline in mild vascular cognitive impairment: the IDEALE study. Clin Interv Aging 8: 131-137.
- Scuderi C, Steardo L (2013) Neuroglial roots of neurodegenerative diseases: therapeutic potential of palmitoylethanolamide in models of Alzheimer’s disease. CNS Neurol Disord Drug Targets 12: 62-69.
- Lilja AM, Luo Y, Yu QS, Röjdner J, Li Y, et al. (2013) Neurotrophic and neuroprotective actions of (-)- and (+)-phenserine, candidate drugs for Alzheimer’s disease. PLoS One 8: 54887.
- Dodel R, Rominger A, Bartenstein P, Barkhof F, Blennow K, et al. (2013) Intravenous immunoglobulin for treatment of mild-to-moderate Alzheimer’s disease: a phase 2, randomised, double-blind, placebo-controlled, dose-finding trial. Lancet Neurol 12: 233-243.
- Contino M, Cantore M, Capparelli E, Perrone MG, Niso M, et al. (2012) A benzopyrane derivative as a P-glycoprotein stimulator: a potential agent to decrease β-amyloid accumulation in Alzheimer's disease. ChemMedChem 7: 391-395.
- Hu S, Cui W, Mak S, Tang J, Choi C, et al. (2013) Bis(propyl)-cognitin protects against glutamate-induced neuro-excitotoxicity via concurrent regulation of NO, MAPK/ERK and PI3-K/Akt/GSK3β pathways. Neurochem Int 62: 468-477.
- Noetze MJ, Gregory KJ, Vinson PN, Manka JT, Stauffer SR, et al. (2013) A novel metabotropic glutamate receptor 5 positive allosteric modulator acts at a unique site and confers stimulus bias to mGlu5 signaling. Mol Pharmacol 83: 835-847.
- Jiang X, Jia LW, Li XH, Cheng XS, Xie JZ, et al. (2013) Capsaicin ameliorates stress-induced Alzheimer’s disease-like pathological and cognitive impairments in rats. J Alzheimers Dis 35: 91-105.
- Tanaka M, Li X, Hikawa H, Suzuki T, Tsutsumi K, et al. (2013) Synthesis and biological evaluation of novel tryptoline derivatives as Indoleamine 2,3-dioxygenase (IDO) inhibitors. Bioorg Med Chem 21: 1159-1165.
- Myrianthopoulos V, Kritsanida M, Gaboriaud-Kolar N, Magiatis P, Ferandin Y, et al. (2013) Novel Inverse Binding Mode of Indirubin Derivatives Yields Improved Selectivity for DYRK Kinases. ACS Med Chem Lett 4: 22-26.
- Dang Z, Jung K, Qian K, Lee KH, Huang L, et al. (2012) Synthesis of Lithocholic Acid Derivatives as Proteasome Regulators. ACS Med Chem Lett 3: 925-930.
- Ereshefsky L, Riesenman C, Lam YW (1995) Antidepressant drug interactions and the cytochrome P450 system. The role of cytochrome P450 2D6. Clin Pharmacokinet 29: 10-18.
- Alderman J, Preskorn SH, Greenblatt DJ, Harrison W, Penenberg D, et al. (1997) Desipramine pharmacokinetics when coadministered with paroxetine or sertraline in extensive metabolizers. J Clin Psychopharmacol 17: 284-291.
- Kuhn UD, Kirsch M, Merkel U, Eberhardt AM, Wenda B, et al. (2007) Reboxetine and cytochrome P450--comparison with paroxetine treatment in humans. Int J Clin Pharmacol Ther 45: 36-46.
- Preskorn SH, Shah R, Neff M, Golbeck AL, Choi J (2007) The potential for clinically significant drug-drug interactions involving the CYP 2D6 system: effects with fluoxetine and paroxetine versus sertraline. J Psychiatr Pract 13: 5-12.
- Liston HL, DeVane CL, Boulton DW, Risch SC, Markowitz JS, et al. (2002) Differential time course of cytochrome P450 2D6 enzyme inhibition by fluoxetine, sertraline, and paroxetine in healthy volunteers. J Clin Psychopharmacol 22: 169-173.
- Caraci F, Crupi R, Drago F, Spina E (2011) Metabolic drug interactions between antidepressants and anticancer drugs: focus on selective serotonin reuptake inhibitors and hypericum extract. Curr Drug Metab 12: 570-577.
- Brøsen K (2004) Some aspects of genetic polymorphism in the biotransformation of antidepressants. Therapie 59: 5-12.
- Whirl-Carrillo M, McDonagh EM, Hebert JM, Gong L, Sangkuhl K, et al. (2012) Pharmacogenomics knowledge for personalized medicine. Clin Pharmacol Ther 92: 414-417.
- Gillman PK (2007) Tricyclic antidepressant pharmacology and therapeutic drug interactions updated. Br J Pharmacol 151: 737-748.
- Murphy GM Jr, Kremer C, Rodrigues HE, Schatzberg AF (2003) Pharmacogenetics of antidepressant medication intolerance. Am J Psychiatry 160: 1830-1835.
- Murphy GM Jr, Hollander SB, Rodrigues HE, Kremer C, Schatzberg AF (2004) Effects of the serotonin transporter gene promoter polymorphism on mirtazapine and paroxetine efficacy and adverse events in geriatric major depression. Arch Gen Psychiatry 61: 1163-1169.
- Grasmäder K, Verwohlt PL, Rietschel M, Dragicevic A, Müller M, et al. (2004) Impact of polymorphisms of cytochrome-P450 isoenzymes 2C9, 2C19 and 2D6 on plasma concentrations and clinical effects of antidepressants in a naturalistic clinical setting. Eur J Clin Pharmacol 60: 329-336.
- Brøsen K (1998) Differences in interactions of SSRIs. Int Clin Psychopharmacol 13: 45-47.
- Baumann P (1998) Care of depression in the elderly: comparative pharmacokinetics of SSRIs. Int Clin Psychopharmacol 13: 35-43.
- DeVane CL, Donovan JL, Liston HL, Markowitz JS, Cheng KT, et al. (2004) Comparative CYP3A4 inhibitory effects of venlafaxine, fluoxetine, sertraline, and nefazodone in healthy volunteers. J Clin Psychopharmacol 24: 4-10.
- Haduch A, Wójcikowski J, Daniel WA (2006) The effect of tricyclic antidepressants, Selective Serotonin Reuptake Inhibitors (SSRIs) and newer antidepressant drugs on the activity and level of rat CYP3A. Eur Neuropsychopharmacol 16: 178-186.
- Spina E, Scordo MG (2002) Clinically significant drug interactions with antidepressants in the elderly. Drugs Aging 19: 299-320.
Citation: Cacabelos R, Torrellas C (2015) Pharmacogenomics of Antidepressants. J Psychiatr Depress Anxiety 1: 001.
Copyright: © 2015 Ramón Cacabelos, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
© 2026, Copyrights Herald Scholarly Open Access. All Rights Reserved!